Design Guidelines for BNA based Oligonucleotide Probes
Artificially modified nucleic acids, such as bridged nucleic acids (BNAs), can be used to increase the thermal stability of probes and primers while maintaining target recognition. Bridged nucleic acids contain key features that allow for this to happen. Rahman et al. in 2005 synthesized and determined the properties of 2’,4’-BNANC molecules. These nucleotide analogs contain an N-O bridged structure which favors sequence selective duplex and triplex formation in BNA/DNA chimeras. Reported duplex and triplex-forming abilities were slightly higher or similar to 2’4’-BNAs. However, observed nuclease resistance was as high as that of S-oligonucleotides.
Figure 1: Design scheme for the synthesis of artificial nucleotide 2'4'-BNANC.
Bridged nucleic acids (BNAs) are artificial bicyclic oligonucleotides that contain a five-membered or six-memberedbridged structure with a “fixed” C3’-endo sugar puckering that is synthetically incorporated at the 2’, 4’-position of the ribose to afford a 2’, 4’-BNA monomer. BNA monomers can be incorporated into oligonucleotide polymeric structures using standard phosphoramidite chemistry. BNAs are structurally rigid oligonucleotides with increased binding affinities and stability. Oligonucleotide modifications are characterized by the presence of one or more bicyclic ribose analogs. The structural similarity to native nucleic acids and the presence of a nitrogen atom within the bicyclic ring leads to very good solubility in water and allows for easy handling of synthetic primers and probes. In contrast to peptide nucleic acids (PNAs) and minor groove binders (MGBs), but similar to LNAs, BNA monomers can be used for both primers and probes in real time quantitative polymerase chain reaction (RT-Q-PCR) assays.
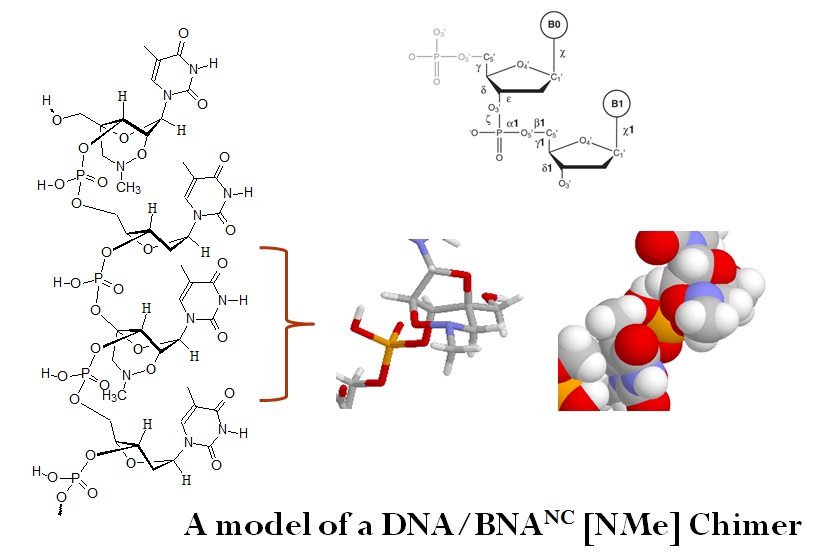
The substitution of DNA monomers with BNA monomers in oligonucleotides adds exceptional biological stability, resistance to nucleases and a significantly increased affinity to their complementary DNA targets. The thermal stability is depending on the number of BNA monomers present in the sequence, and BNA modifications greatly increase the melting temperature of oligonucleotides. Differences in melting temperatures (Tm) between perfectly and imperfectly matched nucleic acid duplexes allow for the discrimination even of single base mutations.
DNA/DNA hybridization based molecular biology techniques rely on the accurate prediction of the melting temperature Tm. Many computer programs using different methods and parameters are available for the theoretical estimation of the experimental Tm value for short oligonucleotide sequences. However, often different values in the estimation of Tm are obtained when using different software packages.
Limitations of Tm estimation calculations are well described in the literature. Ultimately the experimental measurement of Tm values is needed.
BSI now offers the experimental determinations of Tm values for probes and primers as a service.
Panjkovich and Melo, in 2005, compared several different melting temperature calculation methods for short DNA sequences and observed significant differences for the Tm values of short DNA oligonucleotides calculated by different Tm prediction methods. Their results indicate that care needs to be taken when estimating Tm values for oligonucleotides to avoid the failure of PCR experiments.
In the same year, Chavali et al. performed a similar comparative analysis for the prediction of the melting temperature (Tm) for a large set of oligonucleotide sequences. The Tm was predicted using 25 oligonucleotide property calculators. Significant differences in Tm predictions for short DNA sequences were observed when a large number of sequences were tested. Of the 11 primer designing tools evaluated, Primer 3 and WebPrimer performed the best for the AT-rich templates, Exon Primer for AT = GC templates, and Primer Design Assistant, Primer3 and Primer Quest for GC-rich templates.
Conclusion: Many Tm estimation calculation methods do not predict the Tm accurately. Ultimately the experimental measurement of Tm values is needed.
Probe and Primer Design
Tm Optimization Guidelines
Guidelines to help increase the success of practical molecular biology applications when using BNAs, as modeled after Panjkovich and Melo, are listed as follows:
(1) Consider restrictions or limitations that each method has: For example, avoid
sequences that form stable alternative secondary structures because such
sequences do not follow a two-state transition;
(2) If possible, use oligonucleotide sequences that fall in the middle range of
CG-content and are shorter than 20–22mers. This is where most of the current
Tm prediction methods agree;
(3) Avoid the use of sequences that fall in those regions of oligonucleotide feature space
where none of the current methods agrees (see Panjkovich and Melo, 2005);
(4) For large-scale applications with sequences where a two-state transition is not known
to occur, use consensus Tm calculation method as published by SantaLucia et al.,
in 1996.
Tm calculations
Melting temperatures can be calculated using the nearest-neighbor model and thermodynamic data as described by .
The equation used is as follows:
Sums of enthalpy (ΔHd) and entropy (ΔSd) are calculated over all internal nearest-neighbor doublets.
ΔSself is the entropic penalty for self complementary sequences, and
ΔHi and ΔSi are the sums of initiation enthalpies and entropies, respectively.
R is the gas constant (fixed at 1.987 cal/K·mol),
CT is the total strand concentration in molar units and
Tm is the melting temperature given in Kelvin units.
Constant b adopts the value of 4 for non-self-complementary sequences or equal to 1 for duplexes of self-complementary strands or for duplexes when one of the strands is in significant excess.
The thermodynamic calculations assume that the annealing occurs in a buffered solution at pH near 7.0 and that a two-state transition occurs.
Template preparation
Proper sample handling and preparation is needed for every polymerase chain reaction (PCR) method used to monitor gene expression, for quantifying food-borne pathogens, testing viral load, or for any pathogen testing, including forensic analysis and clinical diagnostics. Inhibitors present in the original samples can reduce or even block DNA amplification. Proper pre-PCR processing is needed to avoid PCR inhibition during all steps of the PCR chemistry. To achieve optimal detection limits, high specificity, and short time-to-results, many sample preparation methods have been published in recent years. Many of them combine several sample preparation methods. To achieve detection of very low concentrations of food-borne pathogens, enrichment methods are required.
Sample preparation methods used for PCR can be divided into four major categories, but combination of these methods may also be needed for the detection of low concentration of target sequences:
(1) Biochemical methods
For example, the extraction or purification of RNA or DNA with organic solvent such as phenol-chloroform. DNA and RNA extractions methods have now been automated for many samples. Often, cationic magnetic beads or silica-based filters or suspensions are used to separate nucleic acids from sample matrices.
(2) Culture enrichment methods
These methods involve the cultivation of the target microorganism prior to PCR. The aim is to provide detectable concentrations of target cells prior to PCR.
(3) Immunological methods
Many of these methods are based on magnetic beads coated with antibodies to allow the separation of target cells from their natural environment and to concentrate them further. Immunomagnetic separation methods (IMS) can be used for both, DNA and RNA sample preparation. These have also been automated in recent years. Unfortunately, complex matrices can interfere with immune-capture method and may require further processing such as lysis and additional washing steps, or sometimes even an additional purification step prior to detection.
(4) Physical methods
Examples of physical methods are aqueous two-phase systems, buoyant density centrifugation, flotation, centrifugation or ultracentrifugation, as well as filtration. The success of these methods depends on the physical properties of the target samples.
(5) Combination of methods
The combination of different methods may be needed or can be used to achieve optimal detection. However, combining different methods can be time-consuming and costly. In addition, the KIS (keep it simple) rule should always be applied.
Hydrolysis Probes
A hybridization probe consists of a fragment of DNA or RNA of variable length. In general, hybridization probes are between 100 to 1,000 bases in size. Originally hybridization probes were radioactively labeled. Hybridization probes enable detection of DNA or RNA sequences that are complementary to the probe sequence. These type of probes are now often referred to as “TaqMan probe, Molecular Beacons, Scorpions, or LightCycler probes.
Linear hydrolysis probes consist of oligonucleotides with a fluorescent label on the 5’-end and a quencher molecule on the 3’-end.
(1) Probes are designed to have an annealing temperature above that of the primers.
(2) As the reaction is cooled from the melting temperature above that of the primers,
the probe hybridizes to the target sequence.
(3) Cooling further, the primer hybridizes and the new strand is elongated until the DNA
polymerase reaches the 5’ end of the probe.
(4) The probe is cleaved by 5’-3’ exonuclease activity of the enzyme and the fluorescent
label is released.
(5) Theoretically, a fluorescent label should be released with each amplicon synthesized.
However, in a real experiment, between 4 to 47 % of amplicons are detected when using theses probes. Different probe methods are known to have different sensitivities of detection due to different efficiencies in separating the fluorescent label from the quencher. It has been observed that the insertion of a bridged nucleic acid (BNA/LNA) into a linear hydrolysis probe can increase the detection sensitivity by up to 10-fold.
Scorpion Probes
Scorpion probes offer a different detection system. Scorpion probes combine a forward primer and the detection probe into a single molecule. The fluorophore and quencher are held in close proximity with a stem structure. First, the primer region hybridizes and elongates from the single-stranded target. The Scorpion opens when the template is melted away and the probe region hybridizes to the target region. This hybridization reaction separates the label from the quencher.
BNA based PCR Probes
The composition and sequence context of the selected target sequence for a PCR probe influences the design of the probe. Molecular probes used for diagnostic assay development need to be designed accordingly to allow for the assay to be of high sensitivity and specificity. Certain fundamental rules need to be adhered to when designing an accurate quantitative PCR assay. The composition of primers and probes dictated how well an assay works. For example, the melting temperature of the primer and probe duplex is determined by their sequence and length. The addition of a fluorophore and spacers can also have an influence on the melting temperature. If the nature of the desired target sequence does not support a design according to guidelines the development of a diagnostic assay may be impaired.
BNA-containing oligonucleotides useful for RT-Q-PCR assays can range in length between 12 and 20 nucleotides (nt). This is significantly shorter than unmodified primers and probes displaying the same Tm. Furthermore, primers and probes modified with BNA monomers provide greater flexibility in designing consensus primers and probes for the detection of partially homologous target sequences, such as related viral species and serotypes.
Finally, BNA-modified oligonucleotides in RT-Q-PCR assays reveal a specificity and sensitivity superior to other types of primers and probes.
General guidelines for BNA oligonucleotide design
(1) Add BNA monomers at the site where specificity and discrimination are required.
For example, in allele-specific probes at the SNP position or at the end of allele-
specific primers.
(2) For blocking or clamping probes place one BNA monomer per every 3 bases within
an oligomer. For example in a 20mer oligomer, about 4 or more BNA monomers can
be inserted (there is some degree of freedom as to the exact positions, in other
words, they do not have to be exactly every 3 to 6 bases). According to the specific
application, the mode of BNA modification may need to be changed. Both gapmer- and
chimera-modification with BNA-NC will be effective for antisense application. More than
four continuous natural DNA monomers as part in a BNA-modified oligonucleotide were
found to be necessary to recruit RNase H. For diagnostic application, the modification
of one nucleotide (nt) with a BNA nucleotide may be enough.
(3) The spacing of the BNA monomers need not be different within the oligonucleotide
sequence used for different applications. For example, if the spacing is appropriate
the same BNA monomer may be used.
(4) The use of no more than 4-8 BNA’s within a 20mer probe is recommended but is
depended on the specific application.
(5) Long BNA stretches can have high affinity and interactions between nucleotides
can cause the oligonucleotide to fold onto itself. This can produce a secondary
structure, therefore usingruns of three or more G bases are generally not
recommended.
(6) Avoid BNA:BNA interactions that cause self-complementarity or are complementarity
to other BNA-containing oligonucleotides.
(7) Each BNA–NC monomer increases the Tm by about 4 to 5 degrees Celsius.
Using this information allows estimation of the Tm of the BNA containing
oligonucleotide.
(8) BNA-NC(NMe) modifications add very high nuclease-resistance to the oligonucleotide
(much more than LNA modification). This property is desirable not only for therapeutic
application but also for diagnostic use.
(9) Blocks of BNA near the 3’ end of primers can prevent polymerase activity.
(10) Keep the GC content between 30 to 60%.
General design guidelines for Real-Time qPCR probes
(1) The Tm of dual-labeled probes is typically slightly higher than the primer annealing
temperature, approximately 65 to 70 °C.
(2) The optimal length of BNA modified dual-labeled probes is 14 to 18 nucleotides.
Even shorter more efficient probes may be obtained by careful design.
(3) Maintain Tm values for BNA labeled probes to match the Tm of the corresponding
longer DNA probes.
(4) Start with substituting every third base with BNAs in the central segment of the probe.
Between 4 to 6 BNA substitutes may be required to obtain a useful Tm.
(5) Avoid stretches of more than 3 G DNA or BNA bases.
(6) To enable detection of single-nucleotide mutations (SNPs) select the probe sequence
so that the mutation is located centrally in the probe. Start with a single BNA at the
SNP location, a triplet covering the mutation, or make a short BNA probe.
Do not position the SNP at the very ends of the probe.
(7) Always check for possible secondary structures in the probe and avoid BNA/BNA
interactions.
(8) Position the dual-labeled probe as close as possible to the forward primer.
(9) Avoid G in the 5’-position next to the fluorophore. Guanosine (G) can quench adjacent
fluorophores.
(10) Select the strand with the lowest amounts of Gs in the probe.
(11) Avoid longer stretches of identical nucleotides.
(12) Keep the GC content between 30 to 60%.
(13) To allow for exonuclease cleavage always add one DNA in the 5’-end of the probe.
BNAs in PCR Primers
(1) BNAs in PCR probes can increase the binding affinity and specificity of PCR primers.
For example, at the 3’ end in allele-specific PCR and in the SNP positions in allele-
specific hybridization probes.
(2) However, not all working DNA primer pairs may be enhanced with the addition of BNAs.
(3) BNAs should be considered as a supplemental tool for the design of primer pairs.
(4) Adding BNAs to the primer will increase Tm leading to an increased affinity which allows
the shortening of the primer sequence.
(5) BNAs may be used to adjust the Tm of primers to match PCR-cycling conditions.
(6) BNAs should be added to the 5’ prime region where modifications result in an increase
of binding affinity but will not negatively affect specificity primarily determined by the
3’ end.
(7) BNA spiked primers may allow for the discrimination of highly homologous, alternatively
spliced isoforms.
(8) For allele-specific primers add only one BNA at the 3’end.
(9) Avoid stretches of more than four (4) BNAs.
(10) A typical 18mer may not contain more than 3 or 4 BNAs.
(11) Do not use blocks of BNA at the 3’ end.
(12) Keep the GC content between 30 to 60%.
(13) Avoid stretches of more than 3 G DNA or BNA base.
(14) The Tm of the primer pairs should be nearly equal.
(15) Typical Tms of PCR primers for dual-labeled assays are in the range from 58 to 60°C.
(16) For allele-specific primers, a single BNA should be placed in the terminal 3’ or the
3’-1 position.
BNAs in Blocker Probes – BNA Clamping
(1) Add 3’ phosphate, or better, a C3 spacer, or any other spacer group, to prevent
extension by the polymerase.
(2) For detection blocker or clamping probes, there are no exact design rules. Adjust
the Tm by adding BNA bases to allow the correct allele probe or the primer to
bind with the desired affinity.
(3) For primer-blocking probes position the discrimination site in the 3’ region.
Add BNAs in this region.
(4) For PCR blockers design the probe to allow only the PCR blocker probe binding
to avoid general PCR inhibition.
Other special applications for BNAs
- MicroRNA (miRNA) detection.
- Universal Probe Libraries.
- Special diagnostic PCR.
- Affinity Capture Probes.
Glossary
|
Tm
|
Melting temperature: The melting temperature is the temperature in °C at which 50% of the oligonucleotide and its perfect complement are in a duplex.
|
|
Td
|
Dissociation temperature: The dissociation temperature is the temperature in °C at which 50% of an oligonucleotide and its perfect filter-bound complement are in duplex at the particular salt concentration and total strand concentration.
|
|
ΔH
|
Enthalpy: The Enthalpy change can be calculated by subtracting the bond energies of the product from the bond energies of the reactions.
|
|
ΔS
|
Entropy: The change in entropy is the tendency for randomness in a system. Natural systems have the tendency toward low enthalpy and high entropy.
|
Reference
Sreenivas Chavali, Anubha Mahajan, Rubina Tabassum, Souvik Maiti and Dwaipayan Bharadwaj; Oligonucleotide properties determination and primer designing: a critical examination of predictions.Bioinformatics. Vol. 21 no. 20 2005, pages 3918-3925. doi:10.1093/bioinformatics/bti633
Sung-Kun Kim, Klaus D. Linse, Parker Retes, Patrick Castro, Miguel Castro; Bridged Nucleic Acids (BNAs) as Molecular Tools. Journal of Biochemistry and Molecular Biology Research 2015; 1(3): 67-71
Available from: URL: http://www.ghrnet.org/index.php/jbmbr/article/view/1235/1527
Nolan and Bustin: PCR Technology, Current Innovations. 3rd edition. CRC Press. 2013.
Alejandro Panjkovich and Francisco Melo; Structural bioinformatics - Comparison of different melting temperature calculation methods for short DNA sequences. Bioinformatics. Vol. 21 no. 6 2005, pages 711–722.
SantaLucia, J., Jr, Allawi, H.T., Seneviratne, P.A. 1996; Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry 353555–3562.
http://bioinformatics.oxfordjournals.org/content/21/6/711.long