Epigenetics and the inheritance of epigenetic defects
By Klaus D. Linse
Have you ever wondered if where you grew up and where you lived or live now, what you eat and what you do had or has an influence on your health? Evidence is mounting that this is the case. Our genes seem to have a memory of our past. Research indicates that the nutritional fate of our grant parents may have left their mark on our genes. How can this be? Bee keepers know how to tell a worker bee from a queen bee. But what makes a worker bee look different from a queen bee? It’s her sister. Unlike in humans, royalty is not inherited in honey bees. The honeybee queen bee and worker bees are genetically identical but the queen larvae get feed a special diet – royal jelly – in large quantities and for long periods to make the difference. The worker bees become slaves to the queen. Something is happening on top of the bee’s genome. Alas, the epigenome came to pass. The honeybee’s epigenome has been studied by Lyko et al. recently and the methylome, the methylated genome, of the brains of worker bees and queen bees has been published in 2010. The pictures below show the two different bees and a depiction of the methylome.
 |
 |
| A worker bee |
The methylome |
Figure 1: A picture of a worker bee (left), and an icon of the methylome is depicted here.
In a paper published in the journal Science in the year 1987 Robin Holliday reports that there is plenty of evidence now originating from many different sources that indicating that control of gene expression in higher organisms is related to the methylation of cytosine in DNA. Furthermore, the pattern of methylation appears to be is inherited. Scientists observed that the loss of methylation, for example resulting from DNA damage, will lead to heritable abnormalities in gene expression. Robin Holliday noted that these events may be important in oncogenesis and aging. Robin further proposed that epigenetic defects in germ line cells due to loss of methylation can be repaired by recombination during meiosis but that some maybe transmitted to offsprings.
The term “epigenetics” was coined by Waddington (1942) to refer to the study of the “causal mechanisms” by which “the genes of the genotype bring about phenotypic effects.” Furthermore he stated that “Epigenetics has different meanings for different scientists”. In the mind of a molecular biologist that may involve the study of heritable changes of the DNA that can be observed during mitosis that cannot be explained by changes in DNA sequence. These changes include DNA methylation, histone modification and others. For other scientists epigenetics could refer to interactions of cells and cell products that lead to morphogenesis and differentiation. Simply speaking, epigenetic is the study of genetic effects on the phenotype that are not caused by alteration of the DNA sequence including heritable effects on a genes or chromosomes function that is not accompanied by a change in the DNA sequence. As we will see the notion of epigentics has a long history. However, only in the last 25 years did it become an intensely studied scientific problem waiting to be solved. The figure below shows the explosive increase in publications for epigenetics in general and the epigenetics related to cardiovascular disease from 1995 to 2010. Due to the immense list of publications in the field only some major representative papers will be reviewed here.

Figure 2: Illustration of some epigenetic mechanisms
 |
Epigenetic publications: total number and number related to cardiovascular disease. PubMed Search performed on October 30th 2010 using the search terms (epigenetics OR DNA methylation OR histone modifications) for “epigenetics”; and (cardiovascular OR cardiovascular disease) for “cardiovascular disease”
*2010 data were projected by multiplying the 2009 publication numbers by the 2007–2009 yearly relative increment. As of October 30th 2010, PubMed listed 6,586 publications on epigenetics and 280 on epigenetics & cardiovascular disease. [Source Baccarelli et al., 2010]
|
Figure 3: Increase in research publications covering epigentics since 1995.
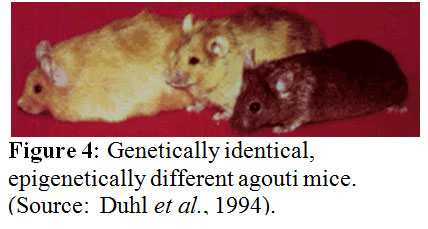 According to Michael J. Meany phenotype emerges only from the interaction of gene and environment. The function of a gene cannot be separated from its cellular environment. Every trait results from the interaction of a gene with the environment. The development of defensive responses to a threat as a maternal effect is a well established theme in biology. Evidence for transgenerational, maternal effects has been published for two animal models – one for a plant and the other for an insect. If a plant gets provoked, say by getting eaten by an insect, it usually reacts with an increase in the synthesis of defensive molecules such as mustard oil glycosides in the case of the radish. These defenses defend against the next attacks. The seedlings derived from caterpillar-damaged radishes showed a significant change in glycosinolate profiles and altered trichrome expression. Since only the mother plants had been exposed to the caterpillars but not the seedlings, these changes have to be adaptive. There are more examples available such as the capacity for night flight in grasshoopers, the tail length of lizards, and the helmet size of water flies and many more are getting discovered the more scientists look for them. These are all determined by maternal effects acting through yet unknown mechanisms. In these examples, traits of the parents are transmitted to the offsprings in a nongenomic manner. The environmental experience of the mother is inherited through an epigenetic mechanism of inheritance causing a phenotypic variation in the offspring. Studies off rats looking for maternal effects revealed that naturally occurring variations in maternal care are associated with individual differences in hypothalamic-pituitary-adrenal (HPA) axis responses to stress. For example the adult offspring of a high licking and grooming mother showed increased hippocampal glucocorticoid receptor expression and enhanced glucocorticoid feedback sensitivity compared to the offspring of a low licking and grooming mother. The first offspring also showed a decrease in hypothalamic corticotrophin-releasing factor (CRF) expression and a more modest HPA response to stress. The result is that the adult offspring rats from frequently licking and grooming mothers are in general behaviorally less fearful and show more modest HPA responses to stress than the offsprings of low licking and grooming mothers. How is this accomplished? In vivo and in vitro studies suggest that the glucocorticoid receptor gene expression is altered through increased hippocampal serotonin (5-HT) activity at 5-HT7 receptors. The increased 5-HT activity results in an increase in the expression of the transcription factor, nerve growth-inducible factor A (NGFI-A) in the hippocampus. The non-coding exon 1 region of the hippocampal glucocorticoid receptor includes a promoter region, exon 17, containing a binding site for NGFI-A. Noncoding regions of the DNA do not code for functional gene products – proteins - and usually contain sequences that regulate the expression of the “downstream” coding segment. For example, exon 1 contains several promoter sequences that can alter gene expression. The exon 17 sequence functions as a promoter in neurons and is more active in the offspring of high licking and grooming mothers, suggesting that the use of this promoter is enhanced as a function of maternal care. Transcription factors such as nerve growth-inducible factor A regulate gene expression and provide a cellular interface between environment and gene. Many details of our behavior and appearance appear to be determined by gene regulation. A striking example of the power of gene regulation is seen in agouti mice (see figure 4), in which genetically identical twins can look entirely different in both color and size. For example, one mouse may be small and brown, but her twin sister may be obese and yellow. Another genetically identical sister may have a mottled look with both fur colors present but may fall in the middle of the weight range. The genome of each of these mice is the same, but the gene expression obviously differs. How can that be? Waterland and Jirtle in 2003 studied the influence of nutrition on adult metabolism in yellow agouti (Avy) mice and showed that research on epigenetic changes resulting from the environment can give clues into obesity in mice--and humans. Their results showed that dietary methyl supplementation of a/a female mice with extra folic acid, B12, choline, and betaine alter the phenotype of their Avy/a offspring via increased CpG methylation at the Avy locus. This locus is a transposable gene element. We conclude, the epigenome is what makes the difference.
According to Michael J. Meany phenotype emerges only from the interaction of gene and environment. The function of a gene cannot be separated from its cellular environment. Every trait results from the interaction of a gene with the environment. The development of defensive responses to a threat as a maternal effect is a well established theme in biology. Evidence for transgenerational, maternal effects has been published for two animal models – one for a plant and the other for an insect. If a plant gets provoked, say by getting eaten by an insect, it usually reacts with an increase in the synthesis of defensive molecules such as mustard oil glycosides in the case of the radish. These defenses defend against the next attacks. The seedlings derived from caterpillar-damaged radishes showed a significant change in glycosinolate profiles and altered trichrome expression. Since only the mother plants had been exposed to the caterpillars but not the seedlings, these changes have to be adaptive. There are more examples available such as the capacity for night flight in grasshoopers, the tail length of lizards, and the helmet size of water flies and many more are getting discovered the more scientists look for them. These are all determined by maternal effects acting through yet unknown mechanisms. In these examples, traits of the parents are transmitted to the offsprings in a nongenomic manner. The environmental experience of the mother is inherited through an epigenetic mechanism of inheritance causing a phenotypic variation in the offspring. Studies off rats looking for maternal effects revealed that naturally occurring variations in maternal care are associated with individual differences in hypothalamic-pituitary-adrenal (HPA) axis responses to stress. For example the adult offspring of a high licking and grooming mother showed increased hippocampal glucocorticoid receptor expression and enhanced glucocorticoid feedback sensitivity compared to the offspring of a low licking and grooming mother. The first offspring also showed a decrease in hypothalamic corticotrophin-releasing factor (CRF) expression and a more modest HPA response to stress. The result is that the adult offspring rats from frequently licking and grooming mothers are in general behaviorally less fearful and show more modest HPA responses to stress than the offsprings of low licking and grooming mothers. How is this accomplished? In vivo and in vitro studies suggest that the glucocorticoid receptor gene expression is altered through increased hippocampal serotonin (5-HT) activity at 5-HT7 receptors. The increased 5-HT activity results in an increase in the expression of the transcription factor, nerve growth-inducible factor A (NGFI-A) in the hippocampus. The non-coding exon 1 region of the hippocampal glucocorticoid receptor includes a promoter region, exon 17, containing a binding site for NGFI-A. Noncoding regions of the DNA do not code for functional gene products – proteins - and usually contain sequences that regulate the expression of the “downstream” coding segment. For example, exon 1 contains several promoter sequences that can alter gene expression. The exon 17 sequence functions as a promoter in neurons and is more active in the offspring of high licking and grooming mothers, suggesting that the use of this promoter is enhanced as a function of maternal care. Transcription factors such as nerve growth-inducible factor A regulate gene expression and provide a cellular interface between environment and gene. Many details of our behavior and appearance appear to be determined by gene regulation. A striking example of the power of gene regulation is seen in agouti mice (see figure 4), in which genetically identical twins can look entirely different in both color and size. For example, one mouse may be small and brown, but her twin sister may be obese and yellow. Another genetically identical sister may have a mottled look with both fur colors present but may fall in the middle of the weight range. The genome of each of these mice is the same, but the gene expression obviously differs. How can that be? Waterland and Jirtle in 2003 studied the influence of nutrition on adult metabolism in yellow agouti (Avy) mice and showed that research on epigenetic changes resulting from the environment can give clues into obesity in mice--and humans. Their results showed that dietary methyl supplementation of a/a female mice with extra folic acid, B12, choline, and betaine alter the phenotype of their Avy/a offspring via increased CpG methylation at the Avy locus. This locus is a transposable gene element. We conclude, the epigenome is what makes the difference.
Historical milestones leading to the notion of epigenetics
| c. 300bc |
Hippocrates thought that humans inherited little parts from all parts of their parents. Whereas Aristotle (around 384-322 bc) thought that humans grew from unformed blobs that developed inside the mother because of the father. Other thoughts of the time were that humans were fully-formed to start with. |
| 1651 |
William Harvey (1578-1657) further explored what Aristotle had put forward. He dissected deer and chicks to understand how embryos form. He came to the conclusion that embryos developed gradually from an egg, rather than from tiny fully-formed bodies.
|
| 1665 |
Cells were described and named by Robert Hooke |
| 1859 |
Concept and facts of evolution were postulated by Charles Darwin |
| 1865 |
In Austria a monk was planting peas and watched how parents passed on their features to the next generation of peas. He established the rules of inheritance, which are the basis of genetics today. His name was Gregor Mendel (1822-1884). Alas the “Mendalian rules” were found in a garden in a monastery in Bruenn (now Brno in the Czech Republic). |
| 1869 |
“Nuclein,” a new acidic, phosphorus-containing, long molecule was found by F. Miescher |
| 1876 |
The notion of “Nature and nurture” was described by F. Galton. |
| 1892 |
August Weissmann (1834-1914) and others recognized that genetic information was stored in the nucleus of a cell. It was thought that cells start with the same information then become more specialized by losing material when they divide. |
| 1902 |
Hans Spemann (1869-1941) agreed with Weissmann but argued that cells don’t lose information; they merely switch it off. He used a strand of his baby’s hair to split a salamander egg in two. The result was two salamanders. Spemann was the pioneer of the modern cloning technology. |
| 1906 |
The term “genetics” was proposed by W. Bateson. |
| 1915 |
It was established that Genes are located on chromosomes leading to the “chromosomal theory of inheritance”. The first genetic linkage in invertebrates was found as well and the term “intersex” was coined. |
| 1926 |
Enzymes are proteins |
| 1942 |
Conrad Waddington (1905-1975) coined the term epigenetics. He thought about development and inheritance in terms of the cross-talk between genetic information and the environment |
| 1953 |
James Watson and Francis Crick (1916- 2004) describe the structure of the DNA double helix in terms of the four letters of the genetic alphabet. DNA is now recognized as the hereditary genetic material. |
| 1980 on |
The gene sequencing revolution begins. Science takes on the view that humans are the sum of their gene sequences. |
| 1990 on |
Epigenetics research takes off. Scientists realize that it is not just DNA sequences which control a human’s biological make-up. DNA methylation and histone modifications are recognized as important regulators of gene activity. |
| 1998 |
RNA interference, RNAi was discovered. Human embryonic stem cells were found. |
2000 to 2001
The first draft sequence of the human genome was published: |
Greg Venter et al. published a paper in Science in 2001 that contained the sequence of the euchromatic portion of the human genome. A 2.91-billion base pair (bp) consensus sequence was generated by the whole-genome shotgun sequencing method. The sequence assemblies covered the euchromatic regions of the human chromosomes. The data showed that more than 90% of the genome is in scaffold assemblies of 100,000 bp or more, and 25% of the genome is in scaffolds of 10 million bp or larger. Analysis of the genome sequence revealed 26,588 protein-encoding transcripts and an additional ∼12,000 computationally derived genes. Almost half the genes are dispersed in low G+C sequence separated by large tracts of apparently noncoding sequences and only 1.1% of the genome is spanned by exons, whereas 24% is in introns, with 75% of the genome being intergenic DNA. Duplications of segmental blocks are abundant throughout the genome. Less than 1% of all SNPs resulted in variation in proteins |
What happens when cells develop into different cell types using the same genome as a template?
Let us first review what we know so far: Human life begins with a single cell. This embryonic “stem cell” contains all the genetic information needed to develop into a full-grown adult, the genome. Through repeated cell divisions the cell eventually multiplies into ten of trillions of cells. Each cell contains a complete copy of the genome. However, despite having the same genetic information these cells develop into hundreds of different cell types that make up the human body. To find out how this works the field of epigenetics came to pass.
A process called mitosis splits a single cell into two cells with identical genetic information. Each cell is capable to develop into different cell types, e.g. into blood cells, neurons or others. We now think that the epigenome determines what type of cell a stem cell will become. Each cell has the same copy of the instruction manual but a brain cell, for example, may only use certain chapters, say to build synapses. The epigenome of each cell tells the cell what chapter to read. Further, we also know now that DNA coils around proteins called histones forming the nucleosome. The helical DNA double strands are packed tightly around the histones inside the nucleus of the cell. The nucleosome is coiled further into a rope like structure called chromatid, which itself is packaged into the chromosome. The epigenome controls access to the genes by attaching molecular caps called methyl groups at certain points of the genes to block them. Histones can coil so tightly around the DNA so that some genes become unreadable. Methyl groups attached to base pairs of a gene changes the expression of the gene. The result is that the DNA of the cells is identical but the epigenetic counterpart is not. Cells perform different functions because now they have different patterns of methyl groups and histones controlling which genes are expressed. DNA methylation is a stable, epigenomic mark occurring at cytosine nucleotides often found within promoter sequences. Histone deacetylases (HDAC) are enzymes that remove acetyl groups (O=C-CH3) from ε-N-acetyl lysines on a histone. The removal of the acetyl group now causes the histones to wrap the DNA around themselves more tightly. DNA methylation is therefore associated with a stable suppression in gene transcription and represents a mechanism to turn genes off.
So what exactly is epigenetics?
Epigenetics is defined as the study of
- Changes in gene expression occurring in organisms with differentiated cells, and the mitotic inheritance of given patterns of gene expression.
- The nuclear inheritance that is not based on changes in DNA sequence.
- "the study of the mechanisms of temporal and spatial control of gene activity during the development of complex organisms." as defined by Robin Holliday.
- The Greek prefix epi- in epigenetics implies features that are "on top of" or "in addition to" genetics; thus epigenetic traits exist on top of or in addition to the traditional molecular basis for inheritance.
- The "epigenome" is used in parallel to the "genome" referring to the overall epigenetic state of a cell. The "epigenetic code" has been used to describe the set of epigenetic features that create different phenotypes in different cells. An epigenomic map would be a representation of gene expression, DNA methylation and histone or other protein modification status of a particular genomic region or the whole genome.
Adrian Bird reports in 2007 in “Perceptions in Epigenetics” that there are two classic epigenetic systems.
A. There is the Polycomb and Trithorax (Polycomb/Trithorax) system, and
B. DNA methylation.
The Polycomb and Trithorax groups of proteins, named after mutants of the fruitfly Drosophila melanogaster, maintain repressed or active transcription states, respectively, of developmentally important genes. If they are absent the genes that specify the different segments of the fruitfly are initially expressed correctly but the gene pattern cannot be maintained. It appears that the Polycomb/Trithorax systems establish stable ’memorized’ gene-expression patterns that have been set up by other cellular mechanisms. It has been reported that Polycomb-imposed silencing can even be transmitted between fruitfly generations at low frequencies. Components of the two key Polycomb-system protein complexes have been identified and a close link with modification of the lysine residue at position 27 of histone H3 has been established. However, the mechanism by which silencing is transmitted between cell generations remains unknown. In vertebrates the methylated sequence is CG, which is paired with the same sequence on the opposite DNA strand. Methylation sites are transiently found on only one of the two DNA strands after DNA replication. CG methylation patterns can be copied between cell generations by the DNA methyltransferase DNMT1, which ‘completes’ hemimethylated but not unmethylated sites. DNA methylation is associated with stable gene silencing (for example, on the inactive X chromosome), either through interference with transcription-factor binding or through the recruitment of repressors that specifically bind sites containing methylated CG. Nuclear receptors can transduce environmental and metabolic signals into alterations in gene expression. The recruited coregulator molecules alter the structure of the chromatin. The following figure shows locations of CpG islands in a hypothetical gene. Methylated and unmethylated islands are depicted.
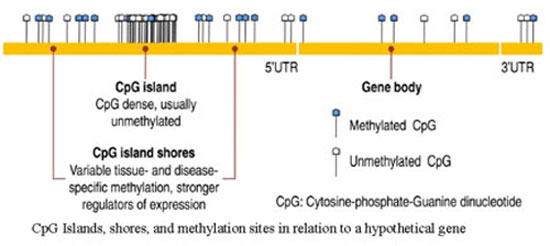
Figure 5: Locations of CpG islands in a hypothetical gene. Methylated and unmethylated islands are shown here.
In the year 2000 Allis and Strahl introduced the notion of the “Histone Code”. The researchers reported that a diverse array of post-translational modifications that often occur on tail domains of histone proteins has been well documented. Furthermore, in their paper they proposed that “distinct histone modifications, on one or more tails, act sequentially or in combination to form a 'histone code' that is read by other proteins to bring about distinct downstream events.” The conventional thinking at the time was that the modifications on the histones just strengthened or loosened the nucleosomes hold on DNA altering gene expression accordingly. The histone code hypotheses proposed instead that these epigenetic modifications represent a histone language that other proteins could read, write and erase and modify. If scientist could decipher this code they could predict events such as transcription, chromatin remodeling and silencing.
In 2001 Jenuwein and Allis published a paper reporting on how to translate the histone code. Chromatin in all eukaryotes contains an array of posttranslational modifications. The majority of which are found on the amino-termini of histones. They proposed that histone proteins and their associated covalent modifications contribute to a mechanism that can alter the structure of chromatin. These modifications then lead to an inherited difference in transcriptional states. They can be viewed as “off” and “on” states that define the higher order structure of the centromers. Chromatin based events can lead to either gene activation or gene silencing. Differences in histone modifications are descriped as “euchromatic” (“on”) or “heterochromatic” (“off”). A schematic representation of the proposed model is depicted in figure 6.
 |
Schematic representation of a model of euchromatin and heterochromatin. Accessible or condensed nucleosome fibers contain different sets of acetylated (Ac), phosphorylated (P), and methylated (Me) histone amino-termini. |
Figure 6: Proposed model of euchromatin and heterochromatin (2001 Jenuwein and Allis) illustrating the “off” and “on” states that define the higher order structure of the centromers.
In the nuclei of all eukaryotic cells, genomic DNA is found as a highly folded, constrained and compacted dynamic polymer. The DNA polymer strands are wound around histones and are compacted further with non-histone proteins. Chromosomal regions that remain transcriptionally inert are highly condensed in the interphase nucleus and can be observed in the cell as heterochromatic foci or as the “Barr body,” which represents the inactive X chromosome in female mammalian cells. Roughly two superhelical turns of DNA wrap around an octamer of core histone proteins in the nucleosome. The core histone proteins that form the octamer are H3-H4, found as a tetramer, and H2A-H2B dimers. Further studies will be needed to find out how the addition of the linker histone 1 (H1) protein causes the chromatin fiber to form a more compacted filament with a defined higher ordered structure. Covalent modifications such as acetylation, methylation, and phosphorylation are found on the histone tails allowing for regulatory contacts with the underlying DNA. Highly specific enzymes that converts these histone tail modifications have been identified and the list of them is getting longer by the day.
The histone code hypothesis predicts that
- distinct modifications of the histone tails will induce interaction affinities for chromation-associated proteins, and
- modifications on the same or different histone tails may be interdependent and generate various combinations on any one nucleosome. Next,
- (distinct qualities of higher order chromatin are largely dependent on the local concentration and combination of differentially modified nucleosomes. It is now thought that this allows a different “read-out” of the genetic information, such as gene activation versus gene silencing, or cell proliferation versus cell differentiation.
Available experimental data link alterations in chromatin structure to cell cycle progression, DNA replication, DNA damage and repair, recombination and chromosome stability.
 |
Examples of combinatorial modifications in the histone amino acid termini that represent imprints for active or inactive chromatin. All four amino terminal ends of the core histones contain short basic patches that often comprise acetylation (Ac), phosphorylation (P), and methylation (Me) marks. |
Figure 7: Examples of combinatorial modifications in the histone amino acid termini representing imprints for active or inactive chromatin.
The field of epigenetics has the potential to revolutionize the field of medical research and healthcare. It encompasses the study of nuclear components such as chromatin structure, including histone modifications, protein/DNA interactions, protein/RNA interactions, and how these factors influence gene function. It also includes the study of DNA methylation and the role that non-coding RNAs (ncRNAs) play in influencing DNA methylation patterns, chromatin structure and the regulation of gene expression. The polymerase chain reaction (PCR) can be used to study DNA methylation patterns, densities, and even the methylation status of individual cytosine residues. In addition, PCR methods have been developed to survey ncRNA expression and to identify regions of the genome where proteins and RNA interact or where certain functional histone marks are located.
References
Adams, J. (2008) Obesity, epigenetics, and gene regulation. Nature Education 1(1)
Adrian Bird. Introduction Perceptions of epigenetics Nature 447, 396-398 (24 May 2007)
Dolinoy, D. C., et al. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proceedings of the National Academy of Sciences 104, 13056–13061 (2007)
Choudhuri S. Obesity, Epigenetics, and Gene Regulation By: Jill U. Adams, Ph.D. (Freelance Science Writer) © 2008 Nature Education
Choudhuri S. From Waddington's epigenetic landscape to small noncoding RNA: some important milestones in the history of epigenetics research. Toxicol Mech Methods. 2011 May;21(4):252-74
Duhl, D. M., et al. Neomorphic agouti mutations in obese yellow mice. Nature Genetics 8, 59–65 (1994).
Andrew P Feinberg. Methylation meets genomics. Nature Genetics 27, 9 - 10 (2001)
D. Haig. “The (Dual) Origin of Epigenetics” 2004, Cold Spring Harbor Symposia on Quantitative Biology Volume LXIX.
Holliday R. The inheritance of epigenetic defects. Science. 1987 Oct 9;238 (4824):163-70.
Holliday, Robin. “Epigenetics: A Historical Overview.” Epigenetics 1(2006): 76-80.
Lu, D., et al. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature 371, 799–802 (1994).
Adele Murrell, Vardhman K. Rakyan and Stephan Beck; From genome to epigenome. Human Molecular Genetics, 2005, Vol. 14, Review Issue 1.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000 Jan 6;403(6765):41-5.
Venter et al., 291 (5507): 1304-1351. The Sequence of the Human Genome Science 16 February 2001: Vol. 291 no. 5507 pp. 1304-1351.
Robert A. Waterland and Randy L. Jirtle. Transposable Elements: Targets for Early Nutritional Effects on Epigenetic Gene Regulation. Mol. Cell. Biol. August 2003 vol. 23 no. 15 5293-5300
Resources for Epigenetics
Consortia and Initiatives
The NIH Roadmap Epigenomics Program: A NIH Initiative to foster epigenomic research, develop comprehensive reference epigenome maps, and generate new technologies for comprehensive epigenomic analyses.
The Epigenome Network of Excellence: An EU-funded network of institutions and research groups
http://www.epigenome-noe.net/WWW/index.php
The Human Epigenome Projects: A public/private collaboration to catalogue Methylation Variable Positions (MVPs) in the human genome
http://www.epigenome.org/
NAME21: A German National Initiative to analyze DNA methylation Patterns of Genes on Chromosome 21
Databases
The Human Epigenome Atlas: The atlas includes human reference epigenomes and the results of their integrative and comparative analyses.
http://www.genboree.org/epigenomeatlas/index.rhtml
MethDB: A searchable database for DNA methylation and environmental epigenetic effects
http://www.methdb.de/
Human Histone Modification Database (HHMD): A searchable database of information from experimental data to facilitate understanding of histone modifications at a systematic level. The current release incorporates 43 location-specific histone modifications in human.
http://bioinfo.hrbmu.edu.cn/hhmd
NCBI Epigenomics: An online repository of epigenetic datasets
GeneImprint: A catalogue of imprinted genes
http://www.geneimprint.com/site/genes-by-species
Catalogue of Parent of Origin Effects: Searchable database of imprinted genes and related effects
Tools and Other Resources
MethPrimer: Primer Design for Methylation PCR
http://www.urogene.org/methprimer/index1.html
MethBlast: A sequence similarity program that checks your primers for bisulfite converted DNA by blasting them against unmethylated and methylated genomic sequences of man, mouse and rat
http://medgen.ugent.be/methBLAST
Methylator: Methylator attempts to predict whether CpGs in a DNA sequence are likely to be methylated or not
RMAP: RMAP is a tool to map reads from the next-generation sequencing technology that supports bisulfite-treated reads mapping.
http://rulai.cshl.edu/rmap/
Chromatin Structure & Function: Information on chromatin biology, histones and epigenetics
http://www.chromatin.us/chrom.html
Epigenetic Station: A source for information, protocols, methods, techniques, products, vendors, kits, assays, analysis, bioinformatics and databases on Epigenetics
http://epigeneticstation.com/
SMART : http://smart.embl-heidelberg.de/