Oligonucleotides that contain a phosphoacetate linkage in place of the standard phosphodiester linkage. Oligonucleotides containing this modification offer improved transfection characteristics, enhanced nuclease resistance, and they show considerable promise in siRNA research. Phosphonocarboxylate oligonucleotides are recently-developed modifications that can be incorporated using “phosphoramidite-like” monomers.
1,2 This phosphonocarboxylate is a broad term that refers to a modified phosphate in which one of the oxygen atoms has been replaced with a carboxylic acid.
PACE Internucleotide Linkage
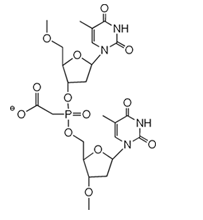
Figure 2: Phosphono Carboxylic Acids
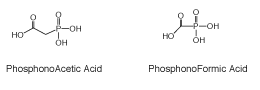
Figure 3: Phosphorothioate and Methyl Phosphonate Internucleotide Linkages
Background
Phosphorus-modified DNA and RNA have been utilized for many years for the biochemical evaluation of DNA/DNA, DNA/RNA, RNA/RNA, protein/RNA, and protein/DNA interactions.5
Phosphorothioates
One of the first chemically modified internucleotide bonds utilized for these studies was phosphorothioate modified oligonucleotide. Methods for the synthesis of phosphorothioate modified DNA were originally developed over 30 years ago by Eckstein in order to probe DNA/protein interactions.6 In the initial rationale for this modification, it was well understood that the negative charge on the internucleotide bond was an important recognition element for the binding of nucleic acids by other nucleic acids and proteins. This modification was particularly appealing because the simple substitution of a sulfur atom for a non-bridging oxygen did not significantly change the crystal structure or charge distribution around small molecule model compounds. Furthermore, the development of efficient and reliable sulfurization protocols7 made the modification as simple to synthesize as normal DNA.
The one significant difference in the physical properties of these molecules compared to normal DNA is the change in hydrogen bonding that is associated with a transformation from oxygen to sulfur. Oxygen can act both as a hydrogen bond acceptor and a hydrogen bond donor; however, sulfur can only act as a weak hydrogen bond donor and cannot act as a hydrogen bond acceptor. Therefore, sulfur substitutions on the phosphodiester internucleotide linkage change the ability of water to shield the negative charge on the phosphodiester through hydrogen bonding interactions. From a practical perspective, this means that phosphorothioate DNA has increased non-specific binding to proteins, which is usually overcome in vitro by increasing salt concentrations.
A second significant difference that accompanies this modification is that a sulfur substitution for oxygen in the non-bridging position of the phosphodiester internucleotide bond generates a stereo center, and the two phosphorus diastereomers show differential binding and recognition by proteins and nucleic acids. Even with these limitations, phosphorothioate modified DNA and RNA have been proven to be very useful tools for biochemistry, molecular biology, cell biology, and oligonucleotide therapeutics.
Methyl Phosphonates
The second significant phosphorus-modified internucleotide bond developed and used for biochemical studies was methylphosphonate DNA (Figure 3). These internucleotide modifications were first proposed by Miller and Tso8 and were specifically intended to address characteristics desirable for therapeutic use of oligonucleotides.
It had been widely observed that highly charged therapeutic molecules, even small molecules like nucleoside mono-phosphates, are not taken up by cells in culture or by animal tissues. The rationale for the methylphosphonate modification was that removing the negatively charged oxygen from the internucleotide bond and replacing it with a non-charged methyl group should increase the ability of these molecules to be taken up by cells in culture and simultaneously protect the resulting modified DNA from being degraded by phosphodiesterases.
What was observed with this modification is that by removing a required recognition element from the DNA mimic, i.e., the negatively charged backbone, the resulting oligonucleotides become inactive in many biological roles. The internucleotide bond in methylphosphonate DNA is highly resistant to nucleases, and reported to give enhanced cellular uptake in various assays. The modification became more broadly accepted when it was demonstrated that it could be incorporated using “phosphoramidite-like” monomers on a typical DNA synthesizer and is now most often incorporated along with natural internucleotide bonds.9
Other Backbones
There have been several additional phosphorus-modified oligonucleotides reported in the scientific literature, most notably phosphorodithioate DNA10, boron phosphonate DNA11, and finally phosphoramidate DNA12. Each of these modifications has demonstrated quite unique and potentially useful structural and biological properties. However, none of these newer modifications has demonstrated significantly enhanced cellular uptake.
Phosphonocarboxylates
Small molecule phosphonocarboxylates, both PAA and PFA (Figure 2), are well known and quite old mimics of phosphate. PFA, as the acid derivative, was originally synthesized over 100 years ago by Arbuzov. Phosphonoformate and Phosphonoacetate are both pretty good mimics of the phosphate group: the 3-dimensional crystal structure is strikingly similar and the charge density distribution is similar. The carboxylic acid functional group of PFA and PAA is effective at hydrogen bonding in both the protonated and charged forms. However, one significant difference is that the ionization constant is changed to that of a weaker acid in the diester form. The diester salt of PAA has a pKa of ≈ 5.0 while the diester salt of phosphate has a pKa of ≈ 2.0. Therefore, when a phosphodiester internucleotide bond is produced with the acetate in the non-bridging position, the protonated and non-protonated forms should be in equilibrium at neutral to mildly acidic pH, Figure 4. This internucleotide bond would be negatively charged, but the charge density would be decreased by the acid/base equilibrium. Asymmetric diesters of phosphonocarboxylates were historically quite difficult to produce. These compounds were typically produced using an Arbuzov reaction, whereby a halide was reacted at elevated temperature with a trialkyl phosphite in a two-step, sequential, oxidative transformation.
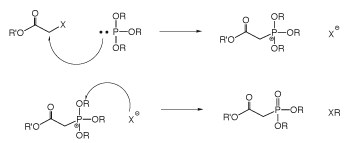
Using this synthetic approach, it was difficult to produce anything but symmetrical diesters. The introduction of phosphinoacetic acid diamidites has significantly changed the synthesis and availability of this class of molecules.3 Using acid activation of phosphonamidites with azole acids (such as tetrazole) phosphonoacetic acid diesters could be produced in greater than 95% stepwise yields; also resulting in the synthesis of phosphonoacetic acid modified DNA.
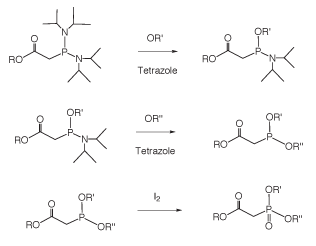
Phosphonocarboxylate Oligonucleotides
The original rationale for the synthesis of phosphonocarboxylate oligonucleotides was to evaluate whether they could be taken into cells through a receptor mediated endocytosis pathway similar to that which had been described for certain carboxylic acid containing glycopeptides.13 It was thought that certain macromolecular glycopeptides are allowed to penetrate into the cytoplasm due to a high concentration of acid neutralizable carboxylate groups on their exterior. These macromolecules would first bind to cell surface receptors and be taken into an endosome. Acidification of the endocytic vesicles after fusion with lysosomes would result in protonation of carboxylic acid chains on the glycopeptides and this pH dependent charge neutralization would in turn allow for penetration of the macromolecule into the cytoplasm. The protonation of carboxylic acid residues that occurs during the acidification of endocytic vesicles has been implicated as a necessary step in various processes including receptor recycling, virus penetration, and the entry of diphtheria toxin into cells.14 The question was whether this proposed mechanism for cellular uptake would function for carboxylic acid modified DNA sequences.
PhosphonoAcetate Oligonucleotides
Mixed sequence phosphonoacetate oligonucleotides were first synthesized and characterized using dimethylcyanoethyl (DMCE) protected phosphinoacetic acid amidites.15 The oligonucleotides were characterized by Sheehan, et. al.16 and shown to have well behaved biochemical properties: they were highly soluble, hybridized in a sequence specific manner to both DNA and RNA, and were highly nuclease resistant.
This existence of the protonated and non-protonated forms in equilibrium on a PACE oligonucleotide was demonstrated by decreasing the pH with mild acid, resulting in greatly increased retention of PACE DNA on a reverse-phase HPLC column. The plot of HPLC retention time as a function of pH gave a sigmoidal curve similar to a titration curve (Figure 5).
A comparison of melting temperatures (ΔTm expressed as a change in °C per linkage) showed a loss of heteroduplex stability for PACE-DNA/DNA and PACE-DNA/RNA relative to natural DNA/DNA and DNA/RNA duplexes, respectively. Losses of approximately 1.3°C per linkage for PACE-DNA/RNA duplexes and approximately 0.3°C for PACE-DNA/DNA duplexes were observed.
The most remarkable observation on PACE DNA was that the fully modified mixed sequence oligonucleotides, 18 to 21 nucleotides in length, demonstrated significantly enhanced uptake with cells in culture.17 There were three specific observations made in these experiments.
First, in comparison to phosphorothioate DNA, or methylphosphonate DNA, a significant amount of PACE DNA was taken up by several cell types in the absence of cationic lipids (all cell types tested). This result was reproducible and verified using Fluorescence Assisted Cell Sorting (FACS) and confocal microscopy. Under these conditions, there was no reproducible cell uptake seen with either fluorescently labelled phosphorothioate DNA or fluorescently labelled methyl phosphonate DNA.
Second, in the presence of cationic lipids, PACE DNA sequences were taken up to a greater extent and with lower lipid concentration than required for normal DNA or phosphorothioate DNA. In most cases, the N to P ratio (the N/P ratio is a measure of the ionic balance of the transfection reagent/DNA complexes, referring to the number of nitrogen residues (ammonium) of the transfection reagent per DNA phosphate) using PACE DNA was allowed to be lowered to the point that little or no cell death could be observed with many cell types while retaining effective transfections.
Third, esterification of the carboxylic acid greatly enhanced the cell uptake of PACE DNA. Remarkably, in order to achieve complete transfections in all cell types evaluated, including JURKAT cells, only 50% of the PACE oligonucleotides needed to be esterified.
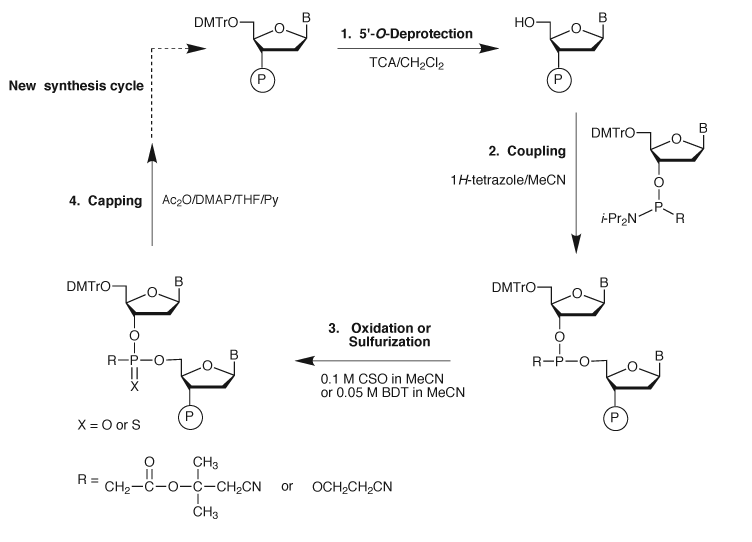
Conclusion
Phosphonoacetate (PACE) modified oligonucleotides show great potential as biological modifiers in a wide variety of research applications. Oligonucleotides containing this modification are easy to synthesize. The monomers can be easily incorporated into complex oligonucleotides and are compatible with a wide variety of other sugar or heterobase modifications. PACE DNA can be conjugated through the carboxylic acid functional group. They have been shown to be active in siRNA duplexes and accelerate the initial rate of cleavage by RNase H-1 when incorporated with phosphorothioates. However, the most interesting observation to date is that they exhibit an unprecedented enhancement in penetration of cultured cells.
- Beaucage, S. L. and Caruthers, M. H., Tetrahedron Lett., 1981, 22 (20), 1859-1862.
- Sinha N. D., Biernat J., McManus J., Köster H., Tetrahedron Lett., 1983, 24 (52), 5843-5846.
- Dellinger, D. J., Sheehan, D. M., Christensen, N. K., Lindberg, J. G., and. Caruthers, M. H., J. Am. Chem.Soc., 2003, 125, 940–950.
- Yamada, C. M.., Dellinger, D. J., and Caruthers, M. H., J. Am.Chem. Soc., 2006, 128, 5251-5261.
- Micklefield, J., Curr. Med. Chem. 2001, 8, 1157–1179.
- Matzura, H., Eckstein, F., Eur. J. Biochem., 1968, 3, 448–452.
- Iyer, R. P., Egan. W., Regan, J. B. and Beaucage, S. L., J. Amer. Chem. Soc., 1990, 112, 1253-1254.
- Miller, P. S., McParland, K. B., Jayaraman, K., Tso, P., Biochemistry, 1981, 20, 1874-1880.
- Hogrefe, R. I., Vaghefi, M. M., Reynolds, M. A., Young, K. M., Arnold L. J., Nucleic Acids Research, 1993, 21, 2031-2038.
- Cummins, L., Graff, D., Beaton, G., Marshall, W. S., Caruthers, M. H., Biochemistry, 1996, 35, 8734–8741.
- Tomasz, J., Ramsay Shaw, B., Porter, K., Spielvogel B. F., Sood, A., Angew.Chem.Int. Ed., 1992, 31, 1373-1375.; Brummel-McCuen, H., Noé, M. S., Sierzchala, A. B., Higson, A. P., Caruthers, M. H., J. Am. Chem. Soc., 2006, 128 (25), 8138-8139.
- Gryaznov, S. M., Skorski, T., Cucco, C., Nieborowska-Skorska, M., Chiu, C. Y., Chen, J. K., Koziolkiewicz, M. Calabretta, B., Proc. Natl Acad. Sci. USA, 1995, 92, 5798–5802.
- Maxfield, F. R., Cell., 1982, 28, 643-651; Tartakoff, A. M., Cell.,1983, 32, 1026-1028.
- Van Renswoude, J., Bridges, K. R., Harford, J. B., Klausner, R. D., Proc. Nail. Acad. Sci. USA ., 1982, 79, 6186-6190; Tycko, B., Keith, C. H., Maxfield, F. R., J Cell Biol.,1983 97(6), 1762-1776.
- Dellinger, D. J., Yamada, C. M., Caruthers, M. H., Current Protocols in Nucleic Acids Chemistry, 2004, 4.24.1-4.24.26.
- Sheehan, D., Lunstad, B., Yamada, C. M., Stell, B., Dellinger, D. J., Caruthers, M. H., Nucleic Acids Research, 2003, 31 (14), 4109-4118.
- Yamada, C. M., Dellinger, D. J., Caruthers, M. H., Nucleosides, Nucleotides, and Nucleic Acids, 2007, 26, 539–54