During aging and disease, cellular damage is accumulated in cells of organisms such as animals and humans. Interstrand DNA crosslinks (ICLs) are formed by metabolic products as well as chemotherapeutic reagents (Muniandy et al. 2010). The maintenance of the genomic integrity of cells is crucial for the survival of all organisms (Ratanaphan, 2012). Damage in the genomic DNA causes disorders, aging, and cancer. A variety of endogenous metabolites, exposure to environmental agents and cancer chemotherapeutics having two reactive groups are known to cause lesions in DNA by covalently joining two DNA strands together. The resulting cross-links are called interstrand cross-links that prevent the separation of the two joined DNA strands. Several synthetic DNA interstrand cross-links have been used for the elucidation of repair pathways (Guainazzi and Scharer, 2010). Synthetic control ICLs are valuable tools for the study of DNA repair pathways for the identification of new therapeutic targets for a more selective cancer chemotherapy.
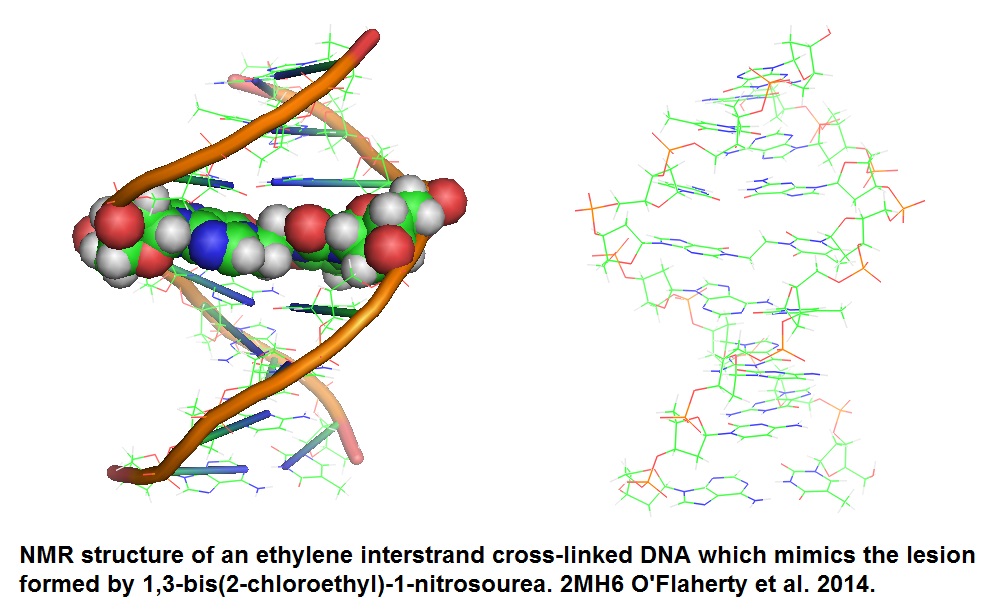
Figure 1: Structure of ethylene ICL DNA (O’Flaherty et al. 2014). The ethylene ICL is highlighted as atomic spheres in the model to the right. This lethal ICL is formed by the bisalkylating agent 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU). BCNU is used in cancer chemotherapy to hinder cellular proliferation. The ethylene linkage ICL connecting the two DNA strands at the N1 atom of 2'-deoxyguanosine and N3 atom of 2'-deoxycytidine is generated by BCNU.
Essential cellular processes including DNA replication and transcription are inhibited by these cross-links. However, it appears to be the job of stem cells to replenish needed tissue cells. It is thought that the damage stem cells sustain over time will compromise their ability to maintain healthy tissues. Information now available about ICLs has increased dramatically during the last decade. For example, Clauson et al. in 2013 reported endogenous sources for DNA ICLs as well as methods describing how to detect, identify and analyze endogenous lesions and repair factors. Table 1 lists common endogenous sources that cause ICLs. Table 2 contains a list of methods useful for the detection and measurement of ICL lesions and their repair.
Table 1: Endogenous sources of DNA interstrand crosslinks.
|
ICL-inducing compounds
|
Target in DNA
|
Synthetic model
|
Endogenous sources
|
|
Aldehydes: Trans-4-
hydroxynonenal, acetaldehyde,
malondialdehyde, acrolein,
formaldehyde, crotonaldehyde
|
5′-GC – nondistorting
5′-CG – distorting
|
Stabilized trimethylene
ICL between two N2-G
|
Lipid peroxidation;
metabolism of dietary
components including:
coffee, ripe fruit and
alcohol
|
|
Nitric oxide, nitrous acid
|
5′-GC < 5′-CG –
distorting
|
Nitrous acid induced
ICL two N2-G
|
Cell signaling;
acidification of dietary
nitrates
|
|
Oxidized abasic lesion: 5′-(2-
phosphoryl- 1,4,dioxobutane)
|
A on the opposite
strand, 3′ to the abasic site
|
Photolabile precursor built into a ss oligonucleotide
|
Hypoxic conditions
|
|
Ring-open aldehyde form of an abasic site 3′ to a C residue
|
G opposite the C
|
AP site built into a ss oligonucleotide; ICL stabilized by reduction with NaCNBH3
|
Spontaneous hydrolysis of
purines or BER repair
intermediates
|
Table 2: Methods to detect and measure ICL lesions and their repair.
|
Method
|
Endpoint Measured
|
Advantages
|
Disadvantages
|
|
Local damage with psoralen + UV-A
|
Covalent addition of psoralen to chromatin in aportion of a cell nucleus
|
Used to identify proteins that co-localize with ICLs to determine the order of events during ICL repair
|
Can only be used in cells cultured in monolayers;
Cannot prove the lesions are ICLs
|
|
Mass spectrometry
|
Levels of mitomycin C or other specific ICL lesions in genomic DNA
|
Highly specific if use
tandem MS; Highly sensitive if used
isotopically labeled
internal standard;
Applicable to cells,
tissues or body fluids
|
Limited to a single lesion per analysis and those for which synthetic standards
are available
|
|
Alkaline COMET assay
|
DNA damage that restricts the electrophoretic mobility of DNA
|
Single-cell measurement of DNA
damage; Sensitive
|
Subjective quantification;
Cannot be used on lesions that are unstable in alkali; Cannot be used on tissues; Genome contains multiple types of DNA damage
|
Note: Both tables are from Clauson et al. (2013).
Recently, scientists presented data that support the idea that stem cells may have intrinsic protective mechanisms that keep them healthy (Katajisto et al. 2015). These recent findings showed that mechanisms exist for mammalianstem like cells to asymmetrically sort aged and young mitochondria. These properties appear to be important to maintain stemness. Stemness is defined as unique characteristics of stem cells which distinguish these cells from normal cells. Stem cells are progenitor or ancestor cells that are able to self-renew and maintain other cells types present in organs. The ability of embryonic (ESCs) and adult tissue stem cells to self-renew and develop into multiple cell lineages is at the heart of the basic definition of “stemness”. Normally, this discovered self-renewal machinery is restricted to stem cells. Therefore one cancer theory suggests that the aberrant activation of the self-renewal machinery in normal cells may be responsible for the initiation of cancer.
Many chemicals are known to produce DNA interstrand cross-links. However, our understanding of the repair of these interstrand cross-links is still incomplete. Research that aims to better understand repair mechanisms for these cross-links is relevant to cancer chemotherapy since tumors often develop resistance to bifunctional anticancer drugs (Noll et al. 2006). Bifunctional anticancer drugs are used in tumor chemotherapy because the tumor tissue growths faster than normal tissue cells and are therefore sensitive to the therapy.
Chemicals that are known to induce interstrand crosslinks are potent genotoxic agents such as bifunctional alkylating agents capable of forming ICLs (Vogel, et al. 1998). Chemotherapy drugs that form ICLs include psoralen and bifunctional alkylating agents such as mechlorethamine, chlorambucil, mitomycin C, and cisplatin. Psoralen induces Thymidine-Thymidine interstrand cross-links (T:T ICLs) induced by irradiation with long-wavelength UV light. However, other compounds react with the nitrogen 7 (N7) on the guanine moiety to form guanidine-guanidine (G:G) ICLs.
ICLs caused by nitrogen mustard are illustrated in figure 2A. Lipid peroxidation products such as acrolein, crotonaldehyde and malondialdehyde which are bifunctional alkylating agents have also been reported to cause ICLs. The cross-links occur at the guanine base. Figure 2B shows the general structure for synthetic ICLs as reported by Guainazzi and Schärer in 2010 and Mukherjee et al. in 2014.
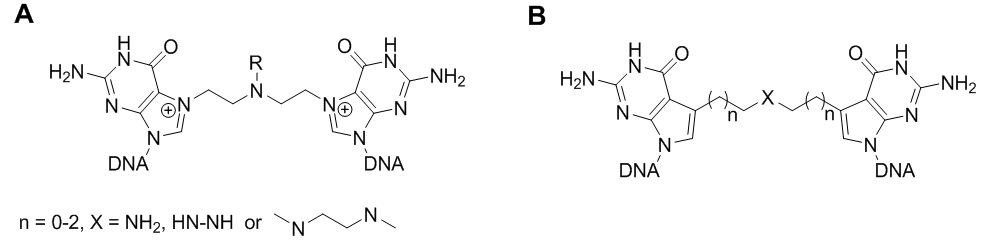
Figure 2. (A) ICLs formed by nitrogen mustard; (B) ICL analogues prepared by Mukherjee et al. 2014 (The Schärer research group).
DNA inter-strand cross-links (ICLs) are toxic DNA lesions and prevent the unwinding of DNA duplex by helicases and thus blocks DNA replication and/or DNA transcription. ICL represent severe DNA lesions, which are highly cytotoxic and genotoxic, and if left intact, can lead to cell death. The repair of ICL lesion has become a hot research topic in the past decade. Unfortunately, the repair mechanisms involved are still not well understood and many biochemical steps involved are still unknown. Nucleotide excision repair (NER) pathways, first discovered in E. coli, are involved in the repair of UV-radiation-induced DNA damage. The base excision repair (BER) pathway and homologous recombination serve to eliminate deleterious lesion from chromosomes and participate in the repair process (Noll et al. 2006). Raeschle et al. in 2008 suggested that nucleotide extension requires DNA polymerase ζ. Furthermore, they argue that a significant portion of the input DNA is fully repaired, but not if DNA replication is blocked. Experiments performed by this research group helped to establish a mechanism for ICL repair that revealed how the repair process is coupled to DNA replication.
A most recent plausible mechanism involves the Fanconi anemia pathway (Deans and West; 2011). Fanconi anemia (FA) is a disorder that is associated with a failure in DNA repair. FA is a genetically heterogeneous recessive disorder characterized by cytogenetic instability, hypersensitivity to DNA cross-linking agents, increased chromosomal breakage, and defective DNA repair. The disorder leads to progressive bone marrow failure and predisposition to cancer. Members of the Fanconi anemia complementation group do not share sequence similarity but are related by how they assembly into a common nuclear protein complex. The gene encodes the protein for complementation group M. Sixteen different genes are known to be involved in Fanconi anemia. The gene products participate in the repair of DNA interstrand cross-links as well as other lesions that block replication fork progression (Coulthard et al. 2013). Eight of the FA proteins assemble in the FA core complex. The protein complex associates with chromatin which leads to the mono-ubiquitination of FANCD2 and FANCI (FANCA-FANCQ). The Fanconi anemia complementation group (FANC) currently includes the proteins FANCA, FANCB, FANCC, FANCD1 (also called BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ (also called BRIP1), FANCL, FANCM and FANCN (also called PALB2). Bogliolo et al. in 2013 reported that mutations in ERCC4, encoding the DNA-repair endonucleose XPF, cause Fanconi Anemia. Biochemical and functional analysis demonstrated that the identified FA-causing ERCC4 mutations strongly disrupt the function of XPF in DNA ICL repair without severely compromising nucleotide excision repair. Whole-exome sequence analysis of blood DNA from one patient revealed a missense mutation in exon 11 (c.2065C > A [p. Arg689Ser]) and a 5 bp deletion in exon 8 leading to a frameshift and premature termination of translation (c.1484_488delCTCAA [p. Thr495Asnfs*6]). Furthermore, sanger sequence analysis of blood DNA from another patient revealed a missense mutation in exon 4 (c.689T > C [p.Leu230Pro]) and a 28 bp duplication in exon 11 leading to a frameshift and a premature stop codon (c.2371_2398dup28 [p.Ile800Thrfs*24]).
TheFANCM protein (defective in FA complementation group M) and the FAAP24 protein complex target other FA proteins to sites of DNA damage. The structure model of the architecture of the DNA recognition elements of the Fanconi anemia FANCM-FAAp24 complex is shown in the following illustration (Figure 3).
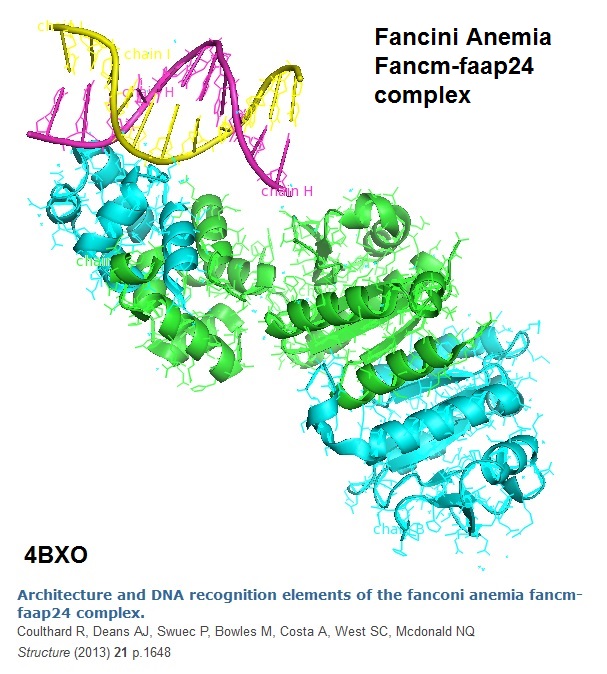
Figure 3: The model of the crystal structure of an S-Shaped FANCMCTD-FAAP24 Complex Bound to dsDNA is depicted (Coulthard et al, 2013).
Model for the ICL Repair Mechanism
Raeschle et al. in 2008 proposed a model for the ICL Repair Mechanism involving the Fanconi anemia pathway based on their experimental data gained from a cell free system base on Xenopus Egg Extracts that support ICL repair. The research group found that during DNA replication of a plasmid containing a site-specific ICL, two replication forks converge on the cross-link. The bypass of the lesions involves the advance of a nascent leading strand to within one nucleotide of the cross-link. Incisions and translesion DNA synthesis followed next. Finally, extension of the nascent strand beyond the lesion occurs. The experimental results suggested that the extension requires DNA polymerase ζ. Unless DNA replication is blocked a significant portion of the cross-linked DNA is fully repaired. The following shows the individual proposed steps involved in the repair mechanism.
A. Formation of Interstrand Cross-link (ICL): A DNA strand containing a nitrogen mustard-like ICL is illustrated in the following figure.
B. Fork Pausing: When the DNA strands are replicated, the leading strands of two converging replication forks initially stall around 20–40 nucleotides (nt) from the lesion. This is called “fork pausing”.
.jpg)
C. Extension to the ICL and possibly replisome remodeling: Next, one leading strand (in red) is then extended to within 1 nt of the ICL (see next figure below). This step is thought to require replisome remodeling. The replisome complex carries out DNA replication starting at the replication origin. This protein complex forms the replication fork and contains several enzymes, including a helicase, a primase, and a DNA polymerase. The replication fork duplicates both DNA strands, the leading and the lagging strand. See the review from Lindås & Bernander (in 2013) for more details.
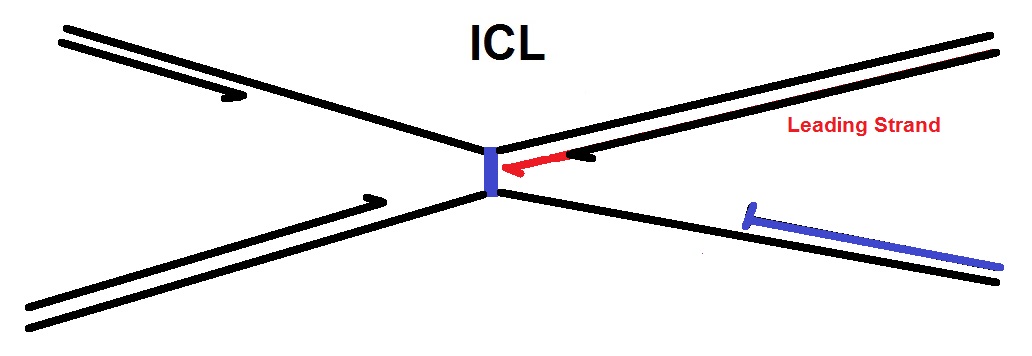
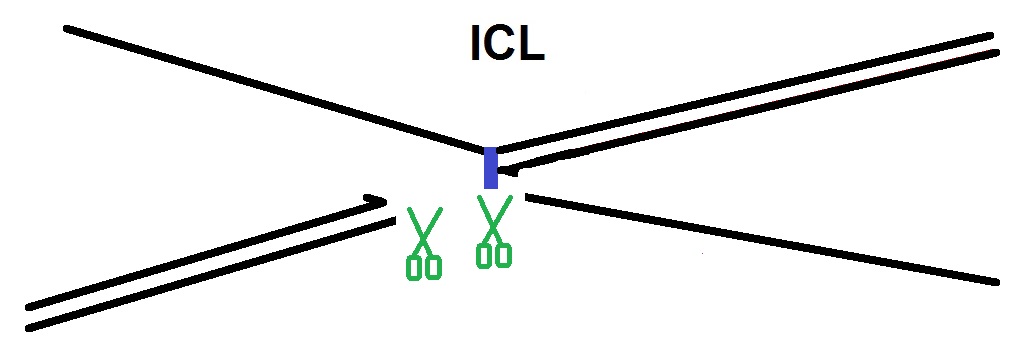
D. In Chromatid uncoupling by incision: In the next step the two sister chromatids that are joined via the ICL are uncoupled via dual incisions on either side of the ICL. The endonucleases that are thought to be involved in the repair process are the endonucleases XPF and Mus81.
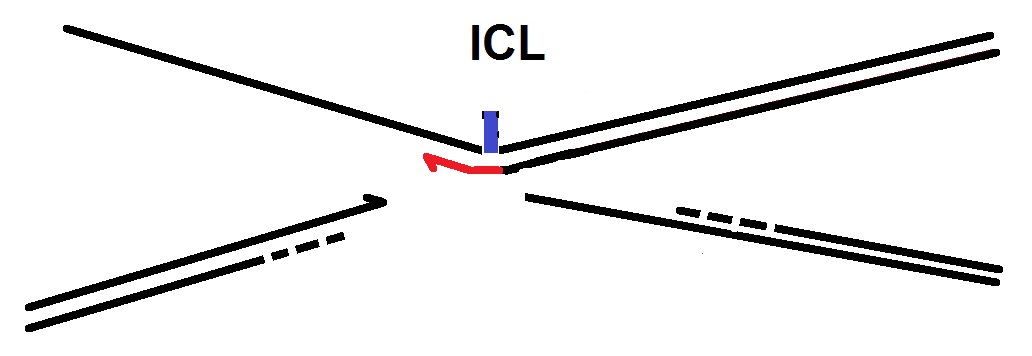
E. Nucleotide insertion: Next, a translesion DNA polymerase, possibly Rev1, inserts a nucleotide across from the adducted base.
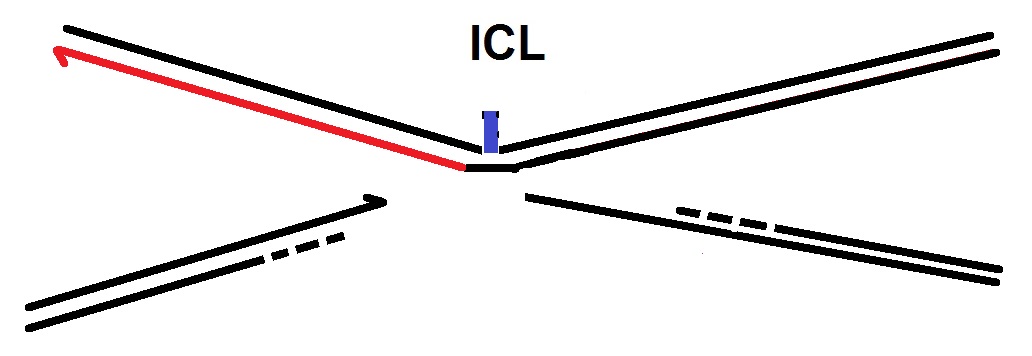
F. Extension past the ICL: In the following step, a DNA polymerase ζ extends the nascent strand beyond the ICL. (Makarova and Burgers, 2015). The DNA polymerase ζ is known to be involved in DNA replication and repair.
G. Repaired DNA Duplexes are generated: Finally, two fully repaired DNA duplexes are generated through the action of nucleotide excision repair (NER) on the top duplex and homologous recombination (HR) on the bottom duplex.
The restriction enzyme Acc I used for the analysis of the repair product recognizes the sequence GT↓(A,C)(T,G)*A*C and generates fragments with 5'-cohesive termini.
However, is has been noted that repair is quite slow. It can take several hours for the repair reaction to complete, in vitro and in mammalian cells.
One of the challenges related to ICL studies at the molecular level is the preparation of DNA duplex substrates that have a single cross-link at a defined site in the duplex. Synthetic ICLs with cross-linked bases site-specifically incorporated into the duplex provides good models for DNA lesion repair and transcription studies as well as mechanism studies of some chemotherapy drugs. However, the preparation of ICLs is difficult because normally two modified bases should be incorporated into the oligonucleotide sequences, and post-synthetic modification is required. A good example is the method published recently by the Schärer group to make ICL mimics that are formed by nitrogen mustard (Figure 1B).
-.-
Bio-synthesis Inc. has over 30 years of experience in oligonucleotide synthesis and offers comprehensive services for customized oligonucleotide synthesis, modification, labeling and conjugation, including the synthesis of inter-strand crosslink analogues upon request.

Reference
Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillón J, Ramírez MJ, Pujol R, Casado JA, Baños R, Rio P, Knies K, Zúñiga S, Benítez J, Bueren JA, Jaspers NG, Schärer OD, de Winter JP, Schindler D, Surrallés J. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013 May 2;92(5):800-6. doi: 10.1016/j.ajhg.2013.04.002. Epub 2013 Apr 25.
Cheryl Clauson, Orlando D. Schärer, Laura Niedernhofer; Advances in understanding the complex mechanisms of DNA interstrand crosslink repair. Cold Spring Harb Perspect Med. Author manuscript; available in PMC 2014 August 6. Published in final edited form as: Cold Spring Harb Perspect Med. 2013 October; 3(10): a012732. PMCID: PMC4123742.
Coulthard R, Deans AJ, Swuec P, Bowles M, Costa A, West SC, McDonald NQ. Architecture and DNA recognition elements of the Fanconi anemia FANCM-FAAP24 complex. Structure. 2013 Sep 3;21(9):1648-58. doi: 10.1016/j.str.2013.07.006. Epub 2013 Aug 8.
Andrew J. Deans, Stephen C. West; DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011 June 24; 11(7): 467–480.
Joseph San Filippo, Patrick Sung, and Hannah Klein; Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008. 77:229–57.
Gene cards: http://www.genecards.org/cgi-bin/carddisp.pl?gene=FANCM
Angelo Guainazzi, Orlando D. Schärer; Using synthetic DNA interstrand crosslinks to elucidate repair pathways and identify new therapeutic targets for cancer chemotherapy. Cell Mol Life Sci. 2010 November; 67(21): 3683–3697.
Pekka Katajisto, Julia Döhla, Christine L. Chaffer, Nalle Pentinmikko, Nemanja Marjanovic, Sharif Iqbal, Roberto Zoncu, Walter Chen, Robert A. Weinberg, and David M. Sabatini; Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 17 April 2015: 340-343. Published online 2 April 2015 [DOI:10.1126/science.1260384] - Stem cells preferentially select new rather than old mitochondria as they divide.
Kim YJ, Wilson DM 3rd.; Overview of base excision repair biochemistry. Curr Mol Pharmacol. 2012 Jan;5(1):3-13.
Ann-Christin Lindås & Rolf Bernander; The cell cycle of archaea. Nature Reviews Microbiology 11, 627-638 (2013). doi:10.1038/nrmicro3077.
Alena V. Makarova, Peter M. Burgers; Eukaryotic DNA polymerase ζ. DNA Repair, Volume 29, May 2015, Pages 47–55.
Mukherjee S, Guainazzi A, Schärer OD; Synthesis of structurally diverse major groove DNA interstrand crosslinks using three different aldehyde precursors. Nucleic Acids Res. 2014 Jun;42(11):7429-35. doi: 10.1093/nar/gku328. Epub 2014 Apr 29.
Parameswary Muniandy, Jia Liu, Alokes Majumdar, Su-ting Liu, and Michael M. Seidman; DNA INTERSTRAND CROSSLINK REPAIR IN MAMMALIAN CELLS: STEP BY STEP. Crit Rev Biochem Mol Biol. 2010 February ; 45(1): 23–49. doi:10.3109/10409230903501819.
David M. Noll, Tracey McGregor Mason, and Paul S. Miller; Formation and Repair of Interstrand Cross-Links in DNA. Chem Rev. 2006 February; 106(2): 277–301
O'Flaherty DK, Denisov AY, Noronha AM, Wilds CJ.; NMR structure of an ethylene interstrand cross-linked DNA which mimics the lesion formed by 1,3-bis(2-chloroethyl)-1-
nitrosourea. ChemMedChem. 2014 Sep;9(9):2099-103. doi: 10.1002/cmdc.201402121. Epub 2014 Jun 16. PMID: 24931822.
Adisorn Ratanaphan; A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer. Review. Int. J. Mol. Sci. 2012, 13(11), 14898-14916; doi:10.3390/ijms131114898
Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC; Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008 Sep 19;134(6):969-80. doi: 10.1016/j.cell.2008.08.030.
Vogel EW, Barbin A, Nivard MJ, Stack HF, Waters MD, Lohman PH.; Heritable and cancer risks of exposures to anticancer drugs: inter-species comparisons of covalent deoxyribonucleic acid-binding agents. Mutat Res. 1998 May 25; 400(1-2):509-40.