The following contains a list of techniques for the analysis of proteins and peptides with protein identification or protein sequence analysis as the final step. Most of these methods have been used and tested for minute sample quantities (> 50 picomoles) in the past decades in many laboratories and by many researchers.
Depending on the solubility of the proteins studied, an extraction and possibly concentration step will be needed before the use of gel electrophoresis, liquid chromatography, ion-chromatography, reversed phase (RP) HPLC, or hydrophobic interaction liquid chromatography (HILIC).
Over the years classical protein biochemistry has evolved into proteomics. Historically, the characterization of unknown proteins began with the extraction and solubilization of selected proteins from tissue samples using an appropriate method or combination of methods. The nature of the protein source determines which extraction method will need to be selected. As many researchers have found out, a complete biochemical characterization of a protein can be quite complex and tricky. Downstream methods used for purification of the protein, generally include affinity chromatography, amino acid analysis, electrofocusing, gel electrophoresis, ion exchange chromatography, mass spectrometry, protein sequencing, as well as size-exclusion or gel filtration chromatography, including spectrophotometry.
Many biochemical methods and assays for the analysis of proteins, peptides, and protein-based products have been developed in the past. To determine the N-terminal end of unknown proteins, Edman based N-terminal protein sequencing is still the method of choice. On the other hand, the availability of more sensitive and highly specific mass spectrometry instruments now allow protein identification at exceedingly lower levels.
For protein identification or proteomic approaches, proteins can be identified and analyzed from:
|
#
|
Sample type
|
Technique or method used
|
|
A
|
Protein(s)
in solution,
or in LC fractions
|
Protein extracts in mixtures are separated using 1D or 2D PAGE, reversed phase HPLC, or in combination with ion-exchange followed by reversed phase HPLC. Fractions containing the protein(s) are collected and enzymatic (e.g. tryptic) in-solution digest is performed. The resulting peptides are analyzed using LC-MS/MS followed by Data Base Searches using a search engine such as Mascot.
Result: -> Internal peptide identification -> List of identified peptides -> Possibly whole sequence coverage of a protein.
|
|
B
|
Gel bands
|
Protein bands are cut out from gels and enzymatic in-gel digest is performed, peptides are extracted and analyzed via LC-MS/MS. Data Searches are perfume using the peptide mass fingerprint patter, for example using the search engine Mascot or similar.
Result -> Internal peptide identification -> List of identified peptides.
When modern methods and instrumentation is used, analysis results can cover the complete sequence of the identified protein(s) including some post-translational modifications.
|
|
C
|
PVDF pieces
|
Protein mixtures are separated using 1D or 2D PAGE, followed by electro-blotting onto a membrane such as PVDF, PVDF piece(s) containing the protein of interest are cut out, and chemical sequencing using Edman Protein Sequencing is performed.
Result: The N-terminal sequence of the protein is observed and reported.
|
|
D
|
PVDF pieces
|
Protein mixtures are separated using 1D or 2D PAGE, followed by electro-blotting onto a membrane such as PVDF, the membrane piece containing the protein is cut out and an enzymatic digest is performed, followed by extracting peptides, the analysis via LC-MS/MS, and Data Base Searches, for example using the search engine Mascot.
Result: -> Internal peptide identification -> List of identified peptides.
|
|
E
|
Lyophylized Proteins
|
Proteins are dissolved in a buffer suitable for downstream analysis. Analysis can be performed using various chromatographic methods as well as gelelectrophoresis as described in A to C.
|
Examples for protein bands in a gel (left) and on a PVDF membrane (right) are shown in figure 1 below. Bands that are as darkly stained may contain between 25 to 50 pm for 50 to 100 kDa proteins. However, bands that only show up as faint bands may contain femtomole amounts of protein. The use of nano-spray LC-MS/MS methods may enable the identification of faint bands as well.
Examples for protein bands in a gel (left) and on a PVDF membrane (right) are shown in figure 1 below. Bands that are as darkly stained may contain between 25 to 50 pm for 50 to 100 kDa proteins. However, bands that only show up as faint bands may contain femtomole amounts of protein. The use of nano-spray LC-MS/MS methods may enable the identification of faint bands as well.
|
|
We recommend performing top-down mass spectrometry on protein or peptide samples after the primary sequence is determined. This type of analysis will confirm the sequence assignment, or, reveal covalent modifications. For example, an observed HexNAc ion (m/z = 204; as observed as a delta mass) may indicate the presence of a glycopeptide (Carr, S.A., Huddleston, M.J., & Bean, M.F. 1993).
A comparison of the observed sequence with the known consensus sequences may help to determine types of modifications present in the protein or peptide (see A. Aitken's Identification of Protein Consensus Sequences). The use of peptide digestions in combination with LC-MS(MS) approaches for assigning modified peptides may also be useful. However, results can be misleading if an unexpected or incomplete digestion occurs, or if the modification is partial or heterogeneous.
Use orthogonal methods during isolation, purification and enrichment steps. For example, we recommend not to use electro-elution twice, rather use electro-blotting or HPLC as the last purification step.
For protein sequencing using Edman chemistry, the higher the protein amount per volume, or per mm2 in the case of a membrane blot, the better the recovery of the protein will be.
|
Abbreviations and nomenclature
|
AAA
|
Amino acid analysis
|
|
Blotting or Electroblotting
|
Electroblotting of proteins and peptides onto polymeric membranes of the PVDF-type
|
|
CE
|
Capillary electrophoresis
|
|
DTT
|
Dithiothreitol
|
|
EC
|
Exclusion chromatography
|
|
HexNAc
|
Hexose N-acetyl
|
|
HIC
|
Hydrophilic ion chromatography
|
|
HPFA
|
Heptafluoroacetone
|
|
HPFP
|
Heptafluoropropanol
|
|
HPLC
|
High performance liquid chromatography
|
|
LC
|
Liquid chromatography
|
|
LC-MS
|
Liquid chromatography-mass spectrometry
|
|
LC-MS/MS
|
Liquid chromatography-tandem mass spectrometry
|
|
Peptide mapping
|
Creating a map of peptide fragments ('finger print') for a protein using chemical or enzymatic digesting methods followed by microseparation methods using either SDS-PAGE, microbore or nanobore HPLC, or capillary or nano-spray LC-MS/MS.
|
|
PVDF
|
Polyvinylidene difluoride
|
|
RPC
|
Reversed phase chromatography
|
|
SDS-PAGE
|
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
|
|
SEC
|
Size exclusion chromatography
|
|
TFA
|
Trifluoroacetic acid
|
|
UV Spectrophotometer
|
Ultra violet spectrophotometer
|
|
XLA
|
Analytical ultracentrifuge
|
Since properties of proteins are different and often protein specific different approaches may need to be designed or developed to achieve success.
Classical methods of protein characterization are generally used for the following type of samples:
- Crude extracts, e.g. from tissues or microbial cells.
- Separation and purification of individual proteins.
- Protein or peptide characterization by determining the molecular mass, the amino acid composition, the protein sequence, as well as potential post-translational modifications.
In general, purification techniques selected can be based on the molecular size of the selected protein. Techniques like dialysis, ultracentrifugation, or size-exclusion chromatography maybe used. Aslo, the solubility of the proteins or peptides of interest may determine which methods to select. Examples are isoelectric point precipitation or salting out methods. If the electric charge of a protein or peptide is known, ion-exchange chromatography or electrophoresis based method maybe selected.
The following flow charts illustrate the use of instrumentation and methodologies for sample preparations of protein and peptides in small or minute amounts. However if higher amounts of protein are available, these protocols can be up scaled as well.
Please note: Microsequencing can be done using the classical approach via Edman based chemical sequencing or by LC-MS/MS based protein or peptide identification methods (proteomics) via mass finger print pattern matching using database searches of genomic data.
A brief description of techniques used for protein analysis, characterization, or proteomics.
|
Technique
|
Result
|
Brief description
|
| |
|
|
|
HPLC
|
Detection of purity of protein as a single peak. A chromatogram is usually reported.
|
Analytical HPLC is used for determination of the protein/peptide purity and to estimate amounts of protein present in a sample. However, the measurement of exact concentrations is not possible unless a control protein with the exact sequence is available for establishing a calibration curve. In addition, HPLC/UV or HPLC/DAD allows the analysis of small organic compounds, peptides, oligonucleotides, antibodies, enzymes, proteins, as well as their conjugates. A variety of reversed-phase columns, including C4, C8, C18, and phenyl-modified columns can be used.
|
|
SDS-PAGE
|
An image of a 1D gradient gel is usually reported with apparent molecular weights.
|
This method removes buffers, lipids, PBS and sugars from the protein.
|
|
Electroblotting
|
An image of the stained membrane containing the protein band(s) is usually reported.
|
Needed for N-terminal Sequencing.
|
|
N-terminal sequencing
|
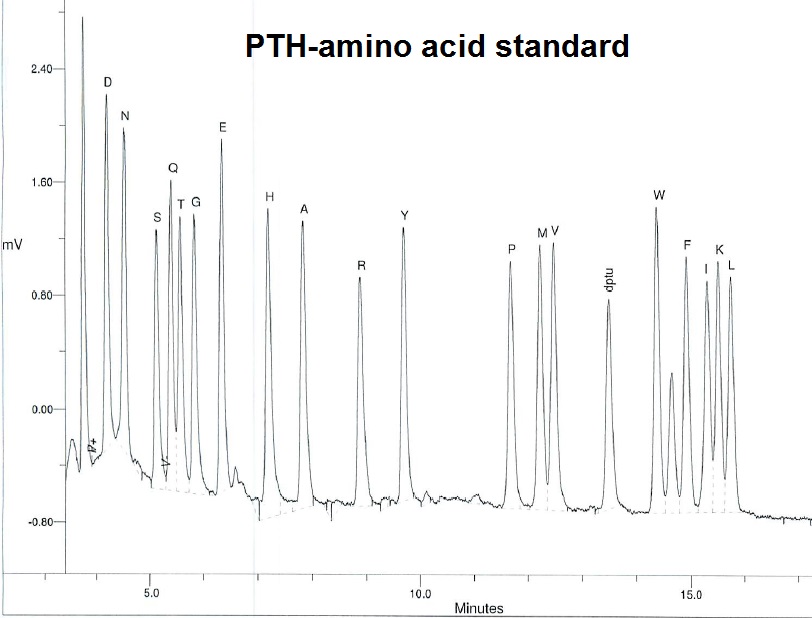
A typical report will contain the observed sequence plus the chromatogram for each cycles of PTH-amino acids released.
|
This method will provide sequence information starting from the N-terminal end of the protein. Often up to 40 or 45 cycles may be observed. However, when the technique was young sequences up to 70 or 80 cycles have been reported.
|
|
UV absorbance at 280 nm
|
The absorbance of the protein at 280 nm is measured and reported. A BSA standard curve is often used to estimate the specific absorbance.
|
Unless the specific absorbance for the protein is known this will only result in an estimate of the protein concentration.
|
|
MALDI-MS
|
This will result in a mass measurement of the protein.
The mass spectra showing the observed peaks are reported.
|
This analysis is more accurate than molecular weight determination via SDS-PAGE. Impurities originating from peptides and protein fragments will be observed as well. Newer instrument types together with accurate calibration can result in highly accurate mass data.
|
|
LC-MS/MS
|
Proteomics. Protein sequence identification is done usually from gel pieces containing the protein of interest via enzymatic (tryptic) digest, followed by LC-MS/MS analysis and database searches.
|
Protein identification via mass pattern matching. The N-terminal and C-terminal end may be present in the data. However it is possible that the N- and C-terminal peptides are missing.
|
|
AAA
|
Determination of protein concentration in a sample. Recovered amino acids are usually reported in picomoles or nanomoles, and nanograms, micrograms, or mgs.
|
Amino acid analysis is still considered to be the most accurate method for determination of protein content. However, matrix effects due to interfering compounds in the formulation buffer are possible. Sometimes the method only works accurately if the protein is highly purified prior to the analysis.
|
Books to review
Aitken's Identification of Protein Consensus Sequences. https://www.ncbi.nlm.nih.gov/nlmcatalog/9103409
Ghon, a. S.; Protein/Peptide Sequence Analysis: Current Methodologies. 1988. CDC Press.
Bollag, D. M., and Edelstein, S. J.; Protein Methods. 1991. Wiley-Liss.
Chen, G.; Characterization of protein therapeutics using mass spectrometry. Springer Science & business Media.
Cutler, P.; Protein Purification Protocols. 2nd edition. Methods in Molecular Biology. Vol 244. Humana Press.
Elzinga, M.; Methods in Protein Sequence Analysis. Humana Press.
Marshak, D., Lin, S-H, Brennan, W.E., Knurth, M., Burgess, R. R.; Strategies for protein purification and characterization. Bookbarn International.
Smith, B. J.; Protein Sequencing Protocols. Methods in Molecular Biology. Vol 211. Humana Press.
Wittman-Liebold, B., Salnikow, J., and Erdman, V. A.; Advanced methods in protein microsequence analysis. Springer Verlag. 1986.
References
Abersold, R.H., Teplow, D.B, Hood, L.E., andKent, S.B.H (1986) J. Bio. Chem. 261:4229-4238.
Aebersold, R.H., Teplow, D.B., Hood, L.E. and Kent St.B.H., (1986); Electroblotting onto activated glass: High efficiency preparation of proteins from analytical SDS-polyacrylamide gels for direct sequence analysis. J. Biol. Chem. 261, (9) 4229-4238.
Abersol, R.H., Leavitt, J., Saavedra, R.A., Hood, L.E. and Kent, S.B.H., (1987) Proc. Natl. Acad. Sci. USA 84:6970-6974.
Aebersold, R.H., Leavitt, J., Hood, L.E. and Kent, S.B.H., (1987); Sequence analysis of proteins from whole cell lysates after separation in analytical two-dimensional gels, p. 277-294; In K. Walsh (Ed.), Methods in Protein Sequence Analysis. Humana Press,NJ.
Bauw, G., De Loose, M., Inzé, D., Van Montagu, M., and Vanderkerckhove, J. (1987) Proc. Natl. Acad. Scie. USA 84:4806-4910.
Bonaventura, C., Bonaventura, J., Stevens, R. and Millington, D. 1994, Anal. Biochem. 222, 44-48. Acrylamide in polyacrylamide gels can modify proteins during electrophoresis.
Carr, S.A., Huddleston, M.J., & Bean, M.F. (1993) Selective identification and differentiation of N- and O-linked oligosaccharides in glycoproteins by liquid chromatography-mass spectrometry. Protein Sci. 2, 183-196. https://www.ncbi.nlm.nih.gov/pubmed/24225996
Hunkapillar and Lujan, in "Methods of Protein Characterization" (JE Shively, ed.) p. 89. Humana Press, Clifton, N.J. (1986).
Kamp, R.M., 1986, High performance liquid chromatography of proteins in Adv. Meth. in Prot. Microseq. Anal. Ed. Wittman-Liebold, Springer 1986; pp. 63-73.
Laemmli, U.K.; Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4 (1970) Nature (London) 227, 680-685. https://www.ncbi.nlm.nih.gov/pubmed/5432063
Matsudaira. P.; Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J. Biol. Chem. 1987 262: 10035-8. http://www.jbc.org/content/262/21/10035.full.pdf+html
M Moos, Jr, N Y Nguyen, and T Y Liu; Reproducible high yield sequencing of proteins electrophoretically separated and transferred to an inert support. J. Biol. Chem. 263:6005 (1988). http://www.jbc.org/content/263/13/6005.full.pdf
Perides, G., Plagens, U., and Traub, P., (1986) Anal. Biochem. Protein transfer from fixed, stained, and dried polyacrylamide gels and immunoblot with protein A-gold. https://www.ncbi.nlm.nih.gov/pubmed/2420231
Renart, J. , Reiser, J., and Stark, G.R., (1979) PNAS 76, 3116-3120. Transfer of Proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: A method for studying antibody specificity and antigen structure. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC383774/
Towbin, H., and Gordon, J., (1984) Journ. Immuno. Meth. 72, 313-340 Immunoblotting and Dot Immunobinding - Current Status and Outlock. https://www.ncbi.nlm.nih.gov/pubmed/?term=Towbin%2C+H.%2C+and+Gordon%2C+J.%2C+(1984)++Journ.+Immuno.+Meth.+72%2C+313-340+Immunoblotting+and+Dot+Immunobinding+-+Current+Status+and+Outlock.
Towbin, H., Staehlin, Th., and Gordon, J., (1979) PNAS 76, 4350-4354. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. https://www.ncbi.nlm.nih.gov/pubmed/388439
Vandekerckhove, J., Bauw, G., Puype, M.,Van Damme, J. & Van Montagu, M. (1985) Protein-blotting on polybrene-coated glass-fiber sheet. Eur: J. Biochem. 152, 9-19. https://www.ncbi.nlm.nih.gov/pubmed/3899644
Wessel and Fluegge, 1984; A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138,141-143. https://www.ncbi.nlm.nih.gov/pubmed/6731838
Wilson, K. J., 1988; Purification of protein/peptides for structural studies in Protein / Peptide Sequence Analysis: Current Methodologies pp. 1-33.
--...---