Chemical conjugation and cross-linking reactions depend on the reactivities of functional groups present in biomolecules and the specificities of conjugation and cross-linking reagents used. In most cases, the selected reaction conditions must preserve the biomolecules' three-dimensional structures to be conjugated or cross-linked. Hence, chemical reagents and reactions need to be chosen that do not inactivate the biological functions of the target molecule. For example, when selecting an amino acid side chain in a protein for conjugation, the amino acid must be located on the protein's surface to be available for the reaction. All proteins are composed of amino acids. The common twenty amino acids have side chains of different sizes, shapes, charges, and chemical reactivity. In the case of proteins, hydrophobic amino acids are generally found in the molecule's interior, forming the hydrophobic core. Amino acids with ionizable side changes are usually located at the protein's surface. Besides amino acids, glycoproteins also contain carbohydrates that provide functional sides for chemical modifications and cross-linking.
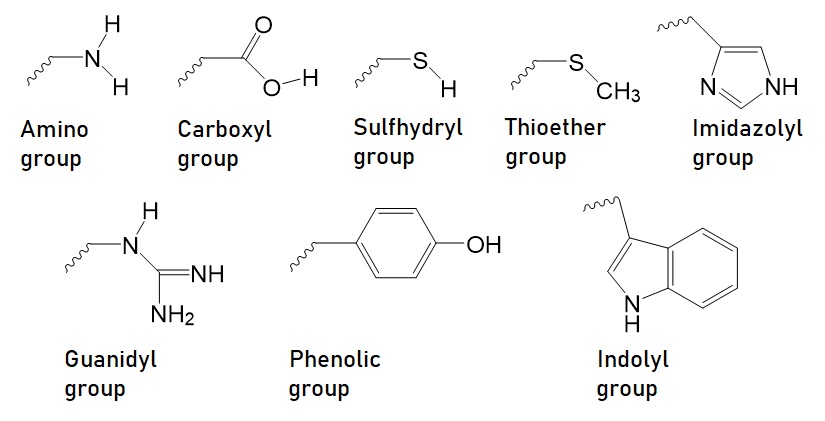
Figure 1: Reactive groups present in biomolecules useful for bioconjugation reaction. The amino, the carboxyl, the sulfhydryl, the thioether, the imidazolyl, and the guanidyl group are the six most reactive functional groups. The phenolic group and the indolyl group are the least reactive functional groups.
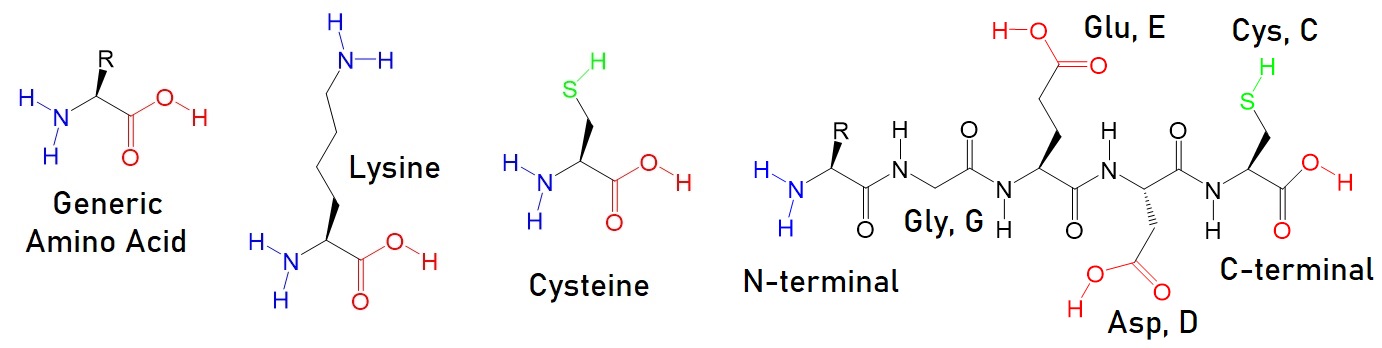
Figure 2: Common amino acid functional groups in peptides and proteins targeted for bioconjugation.
Most of the modifying reactions utilizing these functional groups are nucleophilic reactions. The incoming attacking nucleophile displaces the leaving group. The rate of the biomolecular nucleophilic substitution reaction, the SN2 mechanism, is dependent on at least two factors:
(i) the ability of the leaving group to leave and
(ii) the nucleophilicity of the attacking group.
The greater the nucleophilicity of the incoming group, the faster the product will be formed. The easier the leaving group can come off, the faster the reaction is. The SN2 mechanism of a nucleophilic substitution reaction is illustrated in Figure 3.
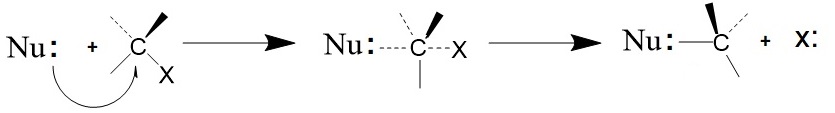
Figure 3: Nucleophilic substitution reaction, the SN2 mechanism. An electron deficient center is attacked by the nucleophile (Nu:) displacing the leaving group (X).
A nucleophile is any molecular species with an unshared pair of electrons, neutral or negatively charged, basically any Lewis base.
The following characteristics govern the nucleophilicity of a species:
[1] A negative nucleophile is always more potent than its conjugate acid. Therefore ArO- is more potent than ArOH,
and OH- is more powerful than H2O.
[2] Nucleophilicity is roughly proportional to the basicity. The approximate order of nucleophilicity is:
NH2- > RO- > OH- >ArO- >RNH2 > NH3 > H2O.
[3] Nucleophilicity increases as one goes down the same column in the periodic table. Hence sulfur is a more powerful
nucleophile than its oxygen analogs.
However, these rules do not always apply. Edwards and Pearson formulated the following order of nucleophilicity:
RS- > ArS- > I- > CN- > OH- > N3- > Br- >ArO- >Cl- >pyridine >AcO- > H2O.
This scheme suggests that the sulfhydryl group of cysteines is the most potent nucleophile in a protein.
Effect of pH
The pH of the medium used for conjugation affects the rate of many nucleophilic reactions because protonation decreases the nucleophilicity of the molecule to be conjugated. The relationship between protonation and the pH is dependent on the pKa of the nucleophile. Table 2 lists the pKa of reactive groups. Since the pKa is a function of temperature, ionic strength, and the micro-environment of ionizable groups, the listed values are only an approximation.
The Henderson-Hasselbalch equation calculates the ratio of protonated to deprotonated species.
pH = pKa + log{[A-]/[AH]}
Based on the equation, the following general rules hold:
[1] At one pH unit below the pKa, the ionizable species is 91% protonated.
[2] At two pH units below the pKa, the ionizable species is 99% protonated.
[3] At one pH unit above the pKa, the ionizable species is 91% deprotonated.
[4] At two pH units above the pKa, the ionizable species is 99% deprotonated.
[5] When the pH is the same as the pKa, 50% protonation occurs.
At a fixed pH, the most reactive group is usually the one with the lowest pKa.
At a neutral pH, the amino groups are protonated and therefore unreactive.
Carboxyl and imidazolyl groups are unprotonated and reactive at a neutral pH.
At pH 5, the imidazolyl group will be over 90% protonated but the carboxyl group will remain in the ionic form.
Other nucleophiles, such as the sulfhydryl group, will react at a higher pH.
Adjusting the pH according to the low pKas allows for controlling the chemical reaction.
Table 1: Chemical modification reactions of amino acid side chains.
|
Amino Acid
|
Side Chain
|
Alkylation or Arylation
|
Acylation
|
Oxidation
|
Others
|
|
Cysteine
|
-CH2SH
|
+
|
+
|
+
|
Iodination, esterification, reaction with mercurial, sulfenyl halides.
|
|
Lysine
|
-NH2
|
+
|
+
|
-
|
Diazotization, amidation, reaction with dicarbonyls.
|
|
Methionine
|
-S-CH3
|
+
|
-
|
+
|
Reaction with cyanogen bromide.
|
|
Histidine
|
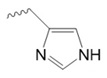
|
+
|
+
|
+
|
Iodination, diazotization.
|
|
Tyrosine
|
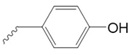
|
+
|
+
|
+
|
Iodination,
Nitration,
Diazotization,
esterification.
|
|
Tryptophan
|
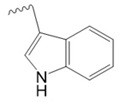
|
+
|
-
|
+
|
Reaction with sulfenyl halides.
|
|
Aspartic and glutamic acids, carboxyl groups.
|
-COOH
|
-
|
+
|
-
|
Esterification, amidation.
|
|
Arginine,
Guanidyl group.
|
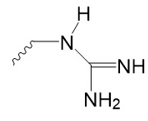
|
-
|
-
|
-
|
Reactions with dicarbonyls.
|
.
Table 2: pKa of reactive groups in biomolecules
|
Functional group
|
AA Residue
|
pKa in free AA
|
pKa in model peptides
|
|
α-COOH
|
C-terminal
|
1.8 – 2.6
|
3.1 – 3.7 (a)
2.1 – 2.4 (b)
|
|
β-COOH
|
Aspartic acid
|
3.9
|
4.4 – 4.6
|
|
γ-COOH
|
Glutamic acid
Aspartic Acid
|
4.3
|
4.4 – 4.6 (a)
4.2 – 45 (b)
3.7 - 4.0 (b)
|
|
.jpg)
|
Histidine
|
6.0
|
6.5 – 7.0 (a)
6.7 – 7.1 (b)
|
|
α-NH3+
|
N-terminal
|
8.8 – 10.8
|
7.6 – 8.0
|
|
-SH
|
Cysteine
|
8.3
|
8.5 – 8.8 (a)
8.8 – 9.1 (b)
|
|
ε-NH3+
|
Lysine
|
10.8
|
10.0 – 10.2 (a)
9.3 – 9.5 (b)
|
|
.jpg)
|
Tyrosine
|
10.9
|
9.6 – 10.0 (a)
9.7 – 10.1 (b)
|
|
.jpg)
|
Arginine
|
12.5
|
>12
|
|
-OH
|
Serine & Threonine
|
>13
|
n.a.
|
Reduction of Disulfide Bonds
In proteins, disulfide bonds cross-link two portions of a protein where cysteines are located. The oxidized form of sulfur is relatively unreactive but can be activated by the reduction of the sulfhydryl group. Thiol-containing reagents such as dithiothreitol, dithioerythritol, 2-mercaptoethanol, and 2-mercaptoethylamine are useful reducing agents.
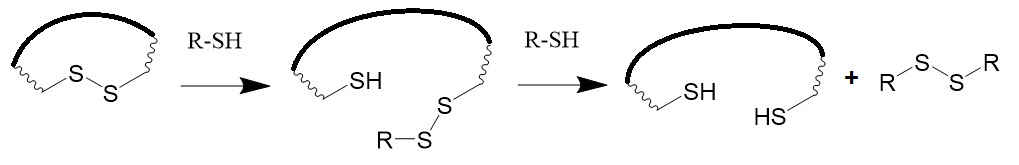
Figure 4: Reduction of disulfide bonds. The reaction involves a disulfide interchange reaction with a free thiol.
The reduction of disulfide bonds is a general reaction used in biochemistry, for example, to prepare immunoglobulin fragments needed for a variety of conjugation reactions.
Interconversion of functional Groups
The introduction of new functional groups by modifying existing functional groups in a biomolecule, such as an amino acid, a peptide, or a protein, allows for a more diverse application of conjugating reactions. For example, it is desirable to convert one functional group into another when preparing immunotoxins.
Conversion of Amines to Carboxylic Acids
The reaction of amino groups with dicarboxylic acid anhydrides will convert these to carboxylic acids. Succinic and malic anhydrides are the two most used dicarboxylic acid anhydrides.
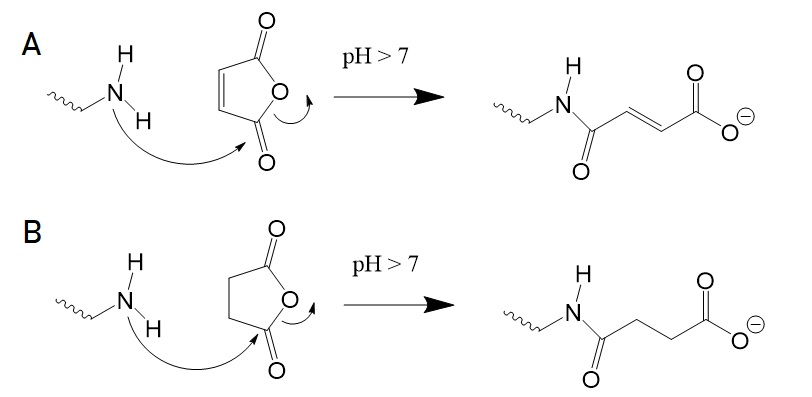
Figure 5: Conversion of amino groups to carboxylic acids. (A) Reaction with malic anhydride. (B) Reaction with succinic anhydride.
Reaction products of maleylation are stable at neutral pH but hydrolyze at acidic pHs. Hence, succinic anhydride is the reagent of choice for chemical conjugation or cross-linking. Succinic anhydride also reacts with tyrosyl, histidyl, cysteinyl, seryl, and threonyl side-chins. However, tyrosyl and histidyl derivatives are reversible. Ester and thioester derivatives are also susceptible to hydroxylamine cleavage at pH 10.
Conjugation of functional groups to amino groups
Several approaches are available to conjugate functional groups or biomolecules to reactive amino groups. Succinic anhydrides, isothiocyanates, and aldehydes allow the linking of functional groups to reactive amino groups.
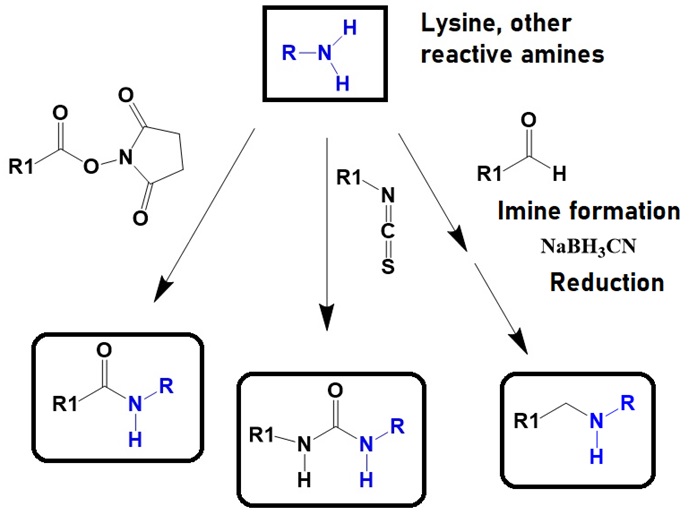
Figure 6: Chemistry of conjugation reactions of reactive amino groups utilizing succinic anhydrides, isothiocyanates, and aldehydes. General chemical structures of the reaction products are shown.
Conversion of amino to sulfhydryl groups
Several approaches are available to link free thiols to amino groups.
(1) Thiolation with N-acetylhomocysteine thiolactone
Nucleophilic attack on the carbonyl group of thiolactones opens the ring, releasing the free thiol group. However, the reaction with amino groups is slow, except at pH 10 to 11. Silver ions catalyze the reaction around pH 7.
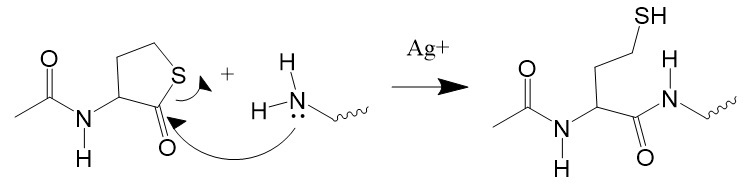
Figure 7: Thiolation with N-acetylhomeocysteine thiolactone.
(2) Thiolation with S-acetylmercaptosuccinic anhydride
In this reaction, the amino group first reacts with the thiol-blocking reagent, followed by treatment with hydroxylamine.
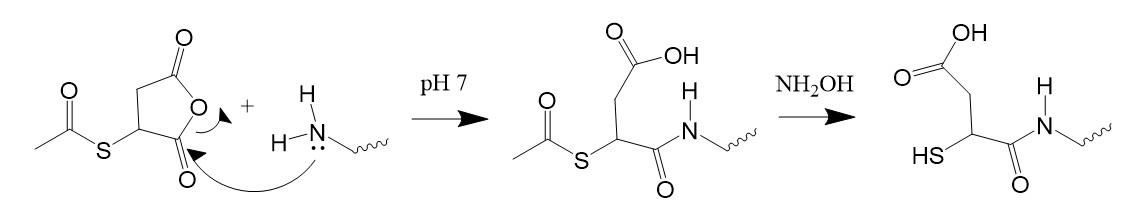
Figure 8: Reaction with S-acetylmercaptosuccinic anhydride.
(3) Thiolation with thiol-containing imidoesters
Several imidoesters allow thiolation of amino groups. Imidoesters react readily with amino groups at pH 7 to 10.
Figure 9: Thiolation with methyl-3-mercaptopropionimidate.
Figure 10: Thiolation with methyl-4-mercaptobutyrimidate.
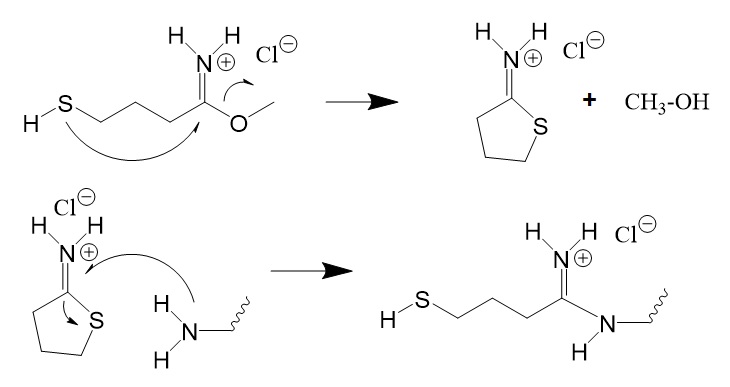

Figure 11: Cyclization reaction of methyl-4-mercaptobutyrimidate to form iminothiolane also called Traut’s reagent followed by its thiolation reaction.
(4) Thiolation with SATA
The succinic anhydride N-succinimidyl S-acetylthioacetate (SATA) allows the addition of sulfhydryl groups to proteins and other amine-carrying molecules such as oligonucleotides. The reaction products can be stored for a long time. Treatment with hydroxylamine exposes the labile sulfhydryl group needed for bioconjugation. The chemical reactions are illustrated in figure.
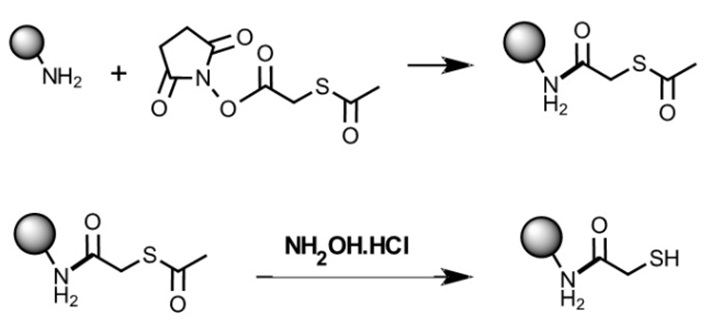
Figure 12: Addition of SATA to amino groups and conversion to free sulfhydryl groups. SATA allows the addition of protected sulfhydryl groups to reactive amines useful for long-time storage. Sulfhydryl groups present in biomolecules, such as oligonucleotides, peptides, and proteins, enable chemical modification of these molecules, for example, for bioconjugation approaches.
Modifications of Carboxyl Groups
Reactions of carboxyl groups with diazomethane, diazoacetate esters and diazoacetamides
(H2C-N2; N2CHC(O)OC2H5; H2N-CO-CH-N2).
Diazomethane, diazoacetate esters, and diazoacetamide react specifically with carboxyl groups of proteins under mild conditions. Diazomethane is protonated by carboxylic acid, forming a carboxylate.
Figure 13: The reaction of diazomethane, diazoacetate esters, and diazoacetate esters with carboxyl groups. The reaction occurs optimally at pH 5. However, diazoacetates and diazoacetamides are highly reactive compounds and react rapidly with water and other inorganic anions reducing their efficiency when reacting with proteins. At a lower pH, hydrolysis is more prevalent.
Reactions of carboxyl groups with carbodiimides
Chemical modification reactions of carboxyl groups using carbodiimide-mediated reactions are the most important conjugation reactions in biotechnology.
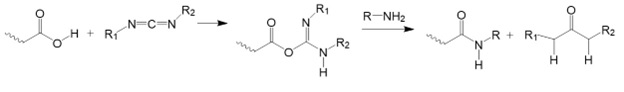
Figure 14: Carbodiimide-mediated modification of carboxyl groups.
Reference
Campodónico PR, Tapia RA, Suárez-Rozas C. How the Nature of an Alpha-Nucleophile Determines a Brønsted Type-Plot and Its Reaction Pathways. An Experimental Study. Front Chem. 2022 Feb 2;9:740161. [PMC]
Edwards J. O., Pearson R. G. (1962). The Factors Determining Nucleophilic Reactivities. J. Am. Chem. Soc. 84, 16–24. [CrossRef]
Hermanson; Bioconjugate Techniques. 3rd edition. Academic Press. 2013. (b)
March’s Advanced Organic Chemistry. 6th edition. 2007, 425-431.; (5) Wong S.S.; Chemistry of protein conjugation and cross-linking. CRC Press. 1993. (a).
Weak acids and bases
---...---
Bio-Synthesis provides a full spectrum of bio-conjugation services including high quality custom oligonucleotide modification services, back-bone modifications, conjugation to fatty acids and lipids, cholesterol, tocopherol, peptides as well as biotinylation by direct solid-phase chemical synthesis or enzyme-assisted approaches to obtain artificially modified oligonucleotides, such as BNA antisense oligonucleotides, mRNAs or siRNAs, containing a natural or modified backbone, as well as base, sugar and internucleotide linkages.
Bio-Synthesis also provides biotinylated mRNA and long circular oligonucleotides.
---...---