BNA PCR clamping enables detection of single point mutations.
Many mutations in human signaling and membrane receptor proteins are the cause of many cancers. Screening for these mutations allows identification of biomarkers that indicate the risk of a patient’s for developing cancer. In addition, after a particular cancer type in a patient has been diagnosed, it is important to screen for resistant of the identified cancer type to therapeutic drugs. For example, the single T790M mutation in the tyrosine kinase domain of the epidermal growth factor receptor (EGRF) allows cancers carrying this mutation to be resistant to gefitinib. Gefitinib is a therapeutic cancer drug used to treat certain breast and lung cancers. Gefitinib inhibits EGRF and interrupts cellular signaling through EGRF in target cells. The cell surface transmembrane glycoprotein EGRF binds to epidermal growth factor. In doing so, it induces receptor dimerization and tyrosine autophosphorylation that leads to cell proliferation. Mutations in this gene are known to be present in lung cancer. Lung cancer is a malignancy that affects lung tissues. The most common forms are divided into three major subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer.
Another example is KRAS or ViKi-ras2 Kirsten rat sarcoma viral oncogene homolog. The KRAS gene is coding for a protein called K-Ras primarily involved in cell division. KRAS is part of the RAS/MAPK pathway in which it relays signals from outside the cell to the cell’s nucleus. The K-Ras protein is an oncogene and a GTPase that acts as a switch that is turned on and off by GTP and GDP molecules. Several mutations in this gene have been identified in people with disorders, and some of them are acquired during a human’s lifetime. Each mutation changes a single amino acid in the protein. Some of these mutations alter chemical signaling in cells throughout the body and can interfere with normal development of many organs or tissues. Mutated oncogenes have the potential to cause cells to become cancerous.
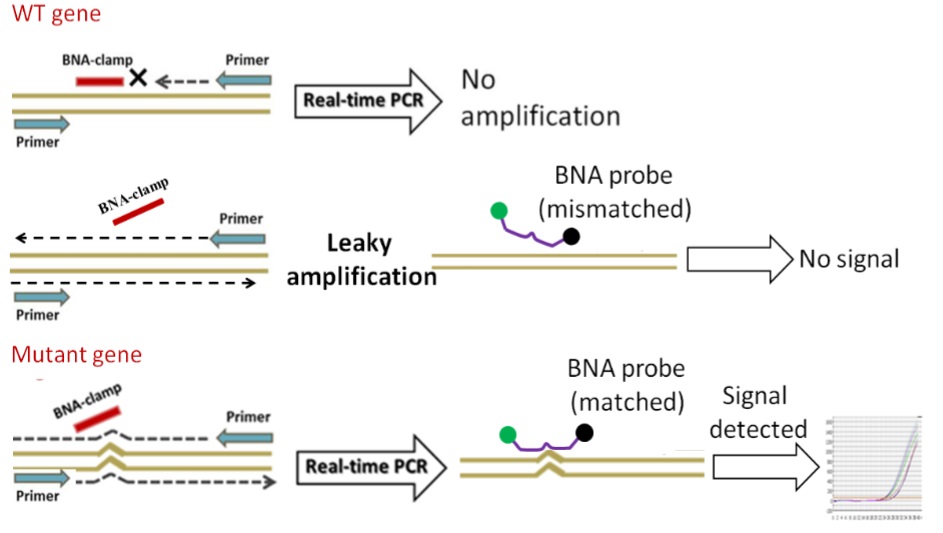
A newly developed bridged-nucleic acid (BNA) based PCR-clamping method or BNA clamping method enables detection of single point mutations such as the T790M mutation in the tyrosine kinase domain of EGRF in a quite simple and cost-effective manner. The BNA clamp selectively suppresses the amplification of the perfect match (wild type) but does not inhibit the amplification of a sequence that differs by as little as one base, for example, mutations. The technique is called PCR Clamping and allows resolving single base differences present in template strands to amplify and detect. This BNA based method allows the selective amplification of many DNA target sequences that differ by a single base pair. The high affinity and specificity of the BNA probe for its complementary nucleic acid sequence prevent the target sequence to function as a primer.
Bridged nucleic acids (BNAs) are known to enhance the hybridization affinity of oligonucleotides that contain BNA bases. BNAclamps are designed to block PCR reaction against wild-type genes to discriminate the presence of mutant genes mixed in with a large number of wild-type genes. The use of Real-Time PCR (RT-PCR) together with BNAclamping allows the detection of different levels of PCR amplifications in mixtures of wild-type and mutant genes.
The polymerase chain reaction, or PCR, is a powerful laboratory technique that allows for the amplification of specific DNA sequences. Since its discovery in 1985, many variations of this method have been introduced and carried out. Today, PCR has become an indispensable part of modern molecular cloning techniques. With the help of PCR, a defined target sequence can be readily and selectively amplified in a quasi-exponential chain reaction. PCR enables the generation of millions of copies, and the method has also been adapted to a wide variety of tasks. PCR is now used in many molecular biology techniques such as DNA sequencing, in vitro mutagenesis, and mutation detection, cloning of cDNA and genomic DNA, as well as allotyping.
However, to get started with any PCR experiment a basic protocol is always a good starting point. Depending on the target DNA modifications to the protocol, may be needed. Because PCR is used for so many applications it is impossible to describe a single set of conditions that will guarantee success in all situations or without further optimizing the protocol for the specific application.
1. Selection of target substrates and PCR primers
The nature of the target DNA together with the specific experiment dictates the nature of the protocol used. The desired substrate DNA should be chosen as clean as possible as well as uncontaminated with other DNA. Select the PCR primers needed.
2. Setting up the reaction
Setting up a PCR requires the following components:
- A thermo stable DNA polymerase to catalyze template-dependent synthesis of DNA.
- A pair of synthetic oligonucleotides to prime DNA synthesis. A forward and reverse primer is needed.
- Deoxynucleoside triphosphate (dNTPs). A standard reaction requires equimolar amounts of dATP, aTTP, dCTP, and dGTP. Recommended concentrations for Taq polymerase are 200 to 250 µM plus 1.5 mM MgCl2 in reaction mixures.
- Divalent cations. Mg, Mn.
- Buffer to maintain pH.
- Monovalent cations. KCl. A standard PCR buffer contains 50 mM KCl. For the amplification of shorter DNA fragments concentrations of ~70 to 100 mM KCl may work better. Circular DNA is amplified slightly less effciently than linear DNAs.
- Template DNA. Target DNA, single- or double-stranded.
In principle PCR can detect a single target molecule, however, in practice several thousand copies of the target DNA are seeded into the reaction mixture.
3. Basic BNA PCR protocol
- Dissolve BNA oligonucleotides in PCR water to a final solution of 100 µM.
- Prepare a stock solution, for example by dissolving 25.44 nmole BNA oligonucleotides in 254.4 µL water.
- Next, dilute 10 µL stock solution in 90 µL PCR water. Use this as a working solution (10 µM).
- Start the PCR using the following protocol:
4. Example: BNA clamping experiment
|
|
Reagent
|
Aliquot
|
|
1
|
2 x SYBR green Mix
|
5 µL
|
|
2
|
10 µM Forward primer
|
0.2 µL (This can be adjusted according to experiment)
|
|
3
|
10 µM Reverse primer
|
0.2 µL (This can be adjusted according to experiment)
|
|
4
|
Target sample (~ 104 copy number or greater)
|
1 µL
|
|
5
|
10 µM BNA clamp
|
1.4 µL (This can be adjusted as needed)
|
|
6
|
H2O
|
2.2 µL
|
|
|
Total PCR reaction volume
|
10 µL
|
5. Real-Time PCR conditions
1 cycle: 95 oC, 10 min; 50 cycles: 95 oC, 15 sec; 60 oC, for 1 min.
This means an initial denaturating step of 10 minutes at 95 °C is followed by 50 cycles of 95 °C for 15 seconds and 60 °C for 1 minute.
Reference
Molecular Cloning: A laboratory manual. 4th edition. Green and Sambrook. Cold Spring Harbor Laboratory Press. 2012.