qPCR is a very useful method for the detection and measurement of bacterial abundance in environmental samples. Using internal standards allows comparison of detected amounts of target amplicon with the internal standards. Internal standards can be prepared either from cloned bacterial DNA or as synthetic DNA blocks using automated solid phase oligonucleotide synthesis. This approach circumvents the problem of differential amplification when using dissimilar targets and standard amplicons. The small subunit ribosomal RNA (SSU rRNA) genes used as target sequences enable the unique identification of bacteria in a sample. Different internal standards are needed to allow estimation of amounts of different bacteria.
In general, PCR typically results in a 105 to 106-fold amplification of target DNA when 20 or 30 PCR cycles are used allowing the design of very sensitive detection and quantification method. The use of general primers amplifies almost all SSU rRNA sequences. However, if unique PCR primers are chosen, only a single species or a small group of bacterial SSU rRNA sequences in the sample will be amplified allowing for selective detection of bacterial species.
PCR is very sensitive which in theory is capable of detecting a single target molecule in samples, but amplification usually saturates during the later cycles. Therefore, the amount of specific sequences in the final amplification product is not always proportional to the amount of the source DNA. Since different sequences can be amplified to different extents, their abundance in the final PCR product does not correlate with their abundance in the original sample; hence quantitative PCR (qPCR) methods are required for the determination of specific SSU rDNA sequences in real samples. Also, the 16S rRNA genes have been used extensively for detection and identification of bacterial species; however, if sequence information on the 23S rRNA gene and the internal transcribed spacer (ITS) region that lies between the 16S and 23S rRNA genes is available, these sequence stretches can also be used as target sequences.
To overcome limitations of sequence discrimination, for example, to prevent the amplification of host sequences when amplifying bacterial DNA, primers containing bridged nucleic acids (BNAs) at specific locations are useful for enhanced amplification of specific target DNA sequences. Another approach is the use of PCR clamping using BNA oligonucleotides for improved and selective amplification of species-specific target DNA.
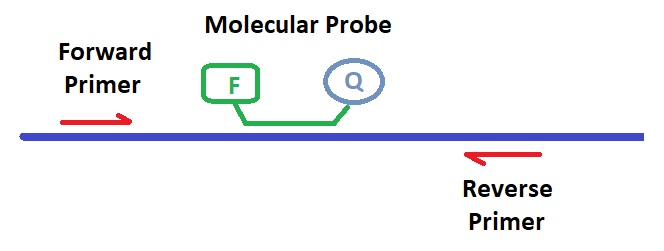
Molecular probes and primer sets enable the qPCR method. FRET-probes that contain a reporter and a quencher molecule, often using a 20 to 30 base pair long oligonucleotide between the fluorophore and the quencher molecule, enabling quantitative PCR. qPCR or TaqManTM probes often contain a 6-FAM reporter dye and a TAMRA quencher; however, other reporter-quencher pairs can be used as well for probe design and synthesis.
Example of how to design and construct a qPCR primer/probe set
• Select the probe and primer to hybridize to a region unique to the targeted DNA with the probe having a Tm that is 10°C higher than the primers.
• An online primer design tool can be used and found at https://molbiol-tools.ca/PCR.htm.
• Design a probe sequence specific for the selected bacterial strain for detection.
• Aim for a sequence in length of 29 base pairs with a target Tm around 70°C.
• Select the reporter dye and quencher, for example 6-carboxyfluorescein (6-FAM) at the 5’-end as the reporter dye and carboxyltetramethylrhodamine (TAMRA) as the quencher at the 3’-end.
• Select the primer sequences such that they flank the probe.
• Select the forward primer be around 25 base pairs in length and to have a Tm of approximately 60°C.
• Select the reverse primer to be approximately 23 base pairs in length and to have a Tm of approximately 59°C.
• Select the primer/probe set to span approximately 90 to 100 base pairs.
• Include a normalizing standard in the PCR assay to correct for any inhibitors present in the sample DNA.
• Use a synthetic oligonucleotide sequence or a cloned oligonucleotide as the internal standard.
• In principle, any sequence not found in the sample is useful as an internal standard.
• Construct a control primer/probe set for the internal standard as well with an alternative fluorochrome as the reporter dye.
• Primers from the normalization sample are selected such that they do not amplify any sequences present in the samples tested.
• The results will be consistent with the composition of the samples tested when sequences in the test samples as well as the internal standard are amplified with both the target primer/probe set and the normalization primer/probe set.
In summary, the design of a qPCR assay involves the selection and construction of internal standardizing sequences and the design of primer pairs plus two fluorescent probes.
Reference
Brunk CF, Li J, Avaniss-Aghajani E.; Analysis of specific bacteria from environmental samples using a quantitative polymerase chain reaction. Curr Issues Mol Biol. 2002 Jan;4(1):13-8. [pubmed].
Ikenaga M, Sakai M. Application of Locked Nucleic Acid (LNA) oligonucleotide-PCR clamping technique to selectively PCR amplify the SSU rRNA genes of bacteria in investigating the plant-associated community structures. Microbes Environ. 2014;29(3):286-95. [pmc].
Si Ming Man, Nadeem O. Kaakoush, Sophie Octavia, Hazel Mitchell; The Internal Transcribed Spacer Region, a New Tool for Use in Species Differentiation and Delineation of Systematic Relationships within the Campylobacter Genus. Appl. Environ. Microbiol. May 2010, 76 (10) 3071-3081; DOI: 10.1128/AEM.02551-09. https://aem.asm.org/content/76/10/3071.
Priya NG, Pandey N, Rajagopal R. LNA probes substantially improve the detection of bacterial endosymbionts in whole mount of insects by fluorescent in-situ hybridization. BMC Microbiol. 2012;12:81. Published 2012 May 24. doi:10.1186/1471-2180-12-81. [pmc].
Friedrich V. Wintzingerode, Ulf B. Göbel, Erko Stackebrandt; Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis, FEMS Microbiology Reviews, Volume 21, Issue 3, 1 November 1997, Pages 213–229, https://doi.org/10.1111/j.1574-6976.1997.tb00351.x, [femsre]
---...---