Abstract: Targeted drug delivery to specific group of cells offers an attractive strategy to minimize the undesirable side effects and achieve the therapeutic effect with a lower dose. Both linear and cyclic peptides have been explored as trafficking moiety due to ease of synthesis, structural simplicity, and low probability of undesirable immunogenicity. Peptides derived from sequence of cell surface proteins, such as intercellular adhesion molecule-1 (ICAM-1), LHRH, Bombesin, and LFA-1, have shown potent binding affinity to the target cell surface receptors. Moreover, peptides derived from ICAM-1 receptor can be internalized by the leukemic T-cells along with the conjugated moiety offering the promise to selectively treat cancers and autoimmune diseases. Systematic analyses have revealed that physicochemical properties of the drug–peptide conjugates and their mechanism of receptor-mediated cellular internalization are important controlling factors for developing a successful targeting system. This review is focused on understanding the factors involved in the development of an effective drug–peptide conjugate with an emphasis on the chemistry and biology of the conjugates. Reported results on several promising drug–peptide conjugates have been critically evaluated. The approaches and results presented here will serve as a guide to systematically approach targeted delivery of cytotoxic drug molecules using peptides for treatment of several diseases.
1. INTRODUCTION
Targeted drug delivery methods have been explored to improve drug efficacy and lower side effects by directing the drug to a specific cell type. To date, many strategies have been investigated to accomplish this goal, and some of these strategies rely on the differences between the cellular compositions of the targeted cells and those of the nontargeted cells. These differences could be in the expressed surface receptors (i.e., the absence or presence of certain receptors), the metabolism profiles (i.e., different enzyme expression or intracellular trafficking), the site/location of the cells (i.e., circulating in blood stream vs. organ), and the nature of the cells (i.e., normal vs. cancerous). For example, tumor cells have certain upregulated receptors, enzymes, and other metabolic features that are not present in normal cells in the body. These differences can be used to discriminate the delivery of a drug to diseased cells rather than to normal cells. Furthermore, the differences in cellular trafficking profiles and the pH of endosomes between normal and cancer cells have also been exploited for selective drug delivery to a specific compartment in the intracellular space of cancer vs. normal cells.1
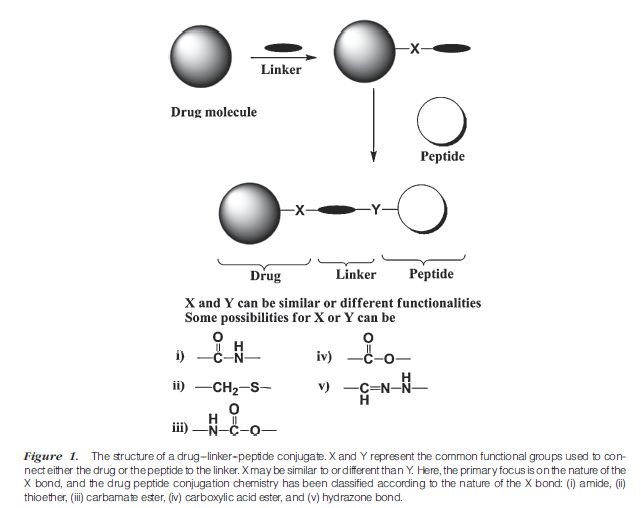
Peptides (e.g., Arg-Gly-Asp (RGD) peptides,2 poly-Arg peptides),3,4 proteins (e.g., antibodies,5 transport proteins, and transferrin6), and small molecules (e.g., folate7) have been used to selectively direct drugs to cancer cells with upregulated receptors by forming drug–carrier conjugates (Fig. 1). However, none of the drug–peptide conjugates has successfully reached the market; this may be due to various factors, including (a) the difficulty in developing the appropriate ligand for the targeted receptor(s) on the cell surface, (b) the lack of understanding of the receptor recycling and trafficking mechanisms, (c) insufficient information on the mechanisms of uptake and disposition of the ligand inside the cells, (d) a lack of understanding of the pharmacokinetic and pharmacodynamic profiles of the drug–peptide conjugate, and (e) the limited number of systematic studies on the relationships between the physicochemical and transport properties of the conjugates. Thus, there is a need to study these aspects of the drug–peptide conjugates to increase the probability of success of these molecules in clinical settings. Ideally, conjugation of the drug to the targeting peptide should not interfere with the recognition of the peptide by its receptor(s). In addition, the
pharmacologic or cytotoxic property of the drug should be maintained when the drug is conjugated to the carrier molecules.
This review illustrates the chemistry, biology, and utilization of small drug–peptide conjugates for selectively delivering the drugs to a specific group of cells (Fig. 1). The first section discusses the attributes of an ideal peptide carrier and the drug. Emphasis will be placed on cyclic peptides and their possible conjugation sites. The second section explores the chemistry behind the drug–peptide conjugation via different types of chemical bonds, including carboxylic acid ester, amide, carbamate ester, hydrazone, and enzymatically cleavable bonds. The presence of a spacer between the drug and the peptide carrier may be necessary to ensure recognition of the carrier by the receptor. The third section focuses on the families of peptides that have been conjugated to drugs and their evaluation in in vitro and in vivo biological systems.
Drug–peptide conjugates are most often administered via the parenteral route because they may not be efficiently delivered orally. The physicochemical properties of peptides (i.e., size, hydrophilicity, and hydrogen-bonding potential) make it difficult for them to cross the intestinal mucosal barriers. In addition, proteases on the surface of intestinal mucosa brush border membranes rapidly metabolize the peptide carrier into fragments. In parenteral delivery, the conjugates travel through the systemic circulation and are distributed to the small capillaries that carry them to the target cells. Upon peptide–receptor interaction, the conjugate can potentially undergo receptor-mediated endocytosis, in which the conjugate may move through the early and late endosomes and finally into the lysosome (Fig. 2). The receptors are either separated from the conjugates in the early sorting endosomes or move to the lysosomes along with the conjugate. After separation from the conjugates, the receptors may be recycled onto the cell surface for the next round of transport. There is a significant drop in pH in the lysosome that may affect the binding between the conjugate and the receptor. It can also affect the linkage between the drug and the peptide (Fig. 2). As an example, the transferrin receptors bind and carry the transferrin protein to transport iron into the cells and, after pH change in the endosomes, the transferrin releases iron and the receptor is recycled back to the cell surface.8 The mechanisms of receptor uptake have been shown to occur via clathrin coat-9 or caveolin-mediated endocytosis (i.e., CD11a/CD18).10
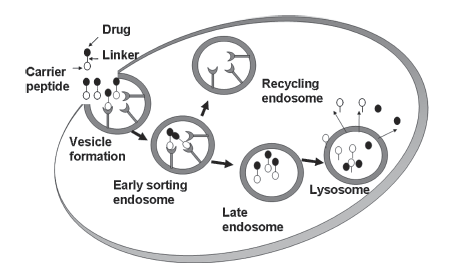
Figure 2. Hypothetical schematic of one possible cellular-internalization process for a drug--peptide conjugate. Drug--linker-- peptide interacts with the cell surface receptor for internalizationvia receptor-mediated endocytosis. Early sorting endosomesmay separate the receptor fromthe conjugate and recycle receptors to cell surface. The conjugate moves to cellular compartments with progressively loweracidity leading to separation of the drug from the carrier. The drug is then released into the cytoplasm to reach the target site for the drug.