Read out probes for smFISH and MERFISH
Transcript analysis in single-cells offers the ability to quantify copy numbers of RNAs and reveal the intercellular RNA location and spatial organization in a variety of cells. Knowing the gene expression in cells is thought to be vital for our understanding of the biology of cells and tissue in different organisms. However, many assays used for the analysis of gene expression destroy the structural context of cells studied.
Advancements made in multiplexed fluorescence in situ hybridization (MERFISH) has increased the analytical throughput tremendously in recent years. For example, Moffit et al. in 2016, reported a measurement throughput for gene profiling of ~40,000 human cell during a period of 18 hours.
Localization of RNAs in cell assemblies or single cells using newer imaging methods provides clues of the spatial distribution and localization of RNAs in normal and disease cells, very important in biomedical research. Newly developed methods such as multiplexed error-robust fluorescence in situ hybridization (MERFISH) now allows studying mRNA molecular biology by using RNA as a reporter molecule. In situ hybridization of fluorescently labeled oligonucleotide probes, as reported by Moffitt and Zhuang, allows quantification of copy number and determination of the spatial distribution of cellular RNA transcripts. MERFISH uses error-robust barcoding for the encoding of RNA species. The barcodes are read out by performing sequential rounds of smFISH measurements.
For smFISH and MERFISH Moffitt et al. designed encoded probes. These probes contained two priming regions, multiple readout sequences, and a target region. For the design of these probes, the GC content and Tm for the target regions in the transcriptome need to be determined. Target sequences for the design of readout probes can be taken from the human transcriptome database, and bioinformatic tools will make the design easier.
Human transcriptome data base: http://useast.ensembl.org/Homo_sapiens/Info/Index.
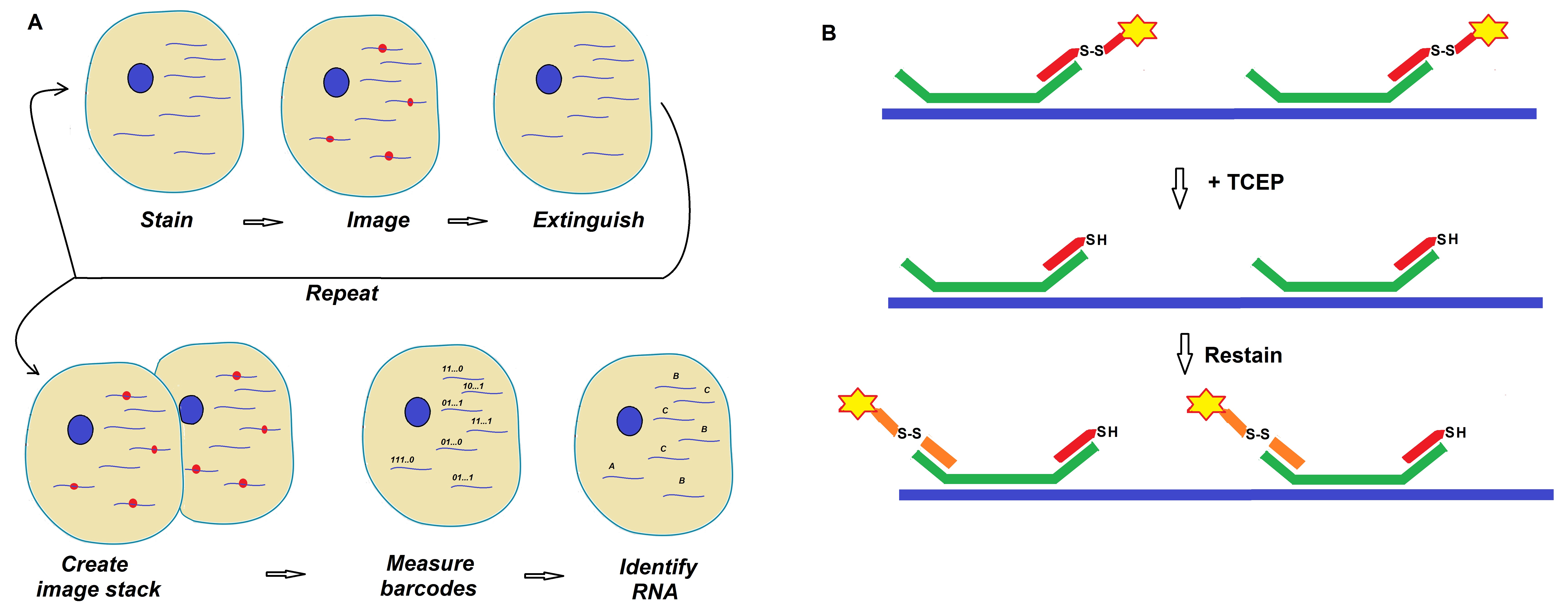
Figure 1: Schematics of a MERFISH readout protocol developed by Moffitt et al. (Moffitt et al. 2016; PNAS).
(A) Target RNAs are stained with encoded probes that contain a barcode and a readout sequence unique to each RNA target. The barcode is identified via successive rounds of smFISH. A stack of images for each sample produces fluorescence spots with on/off patterns that define the barcodes allowing for identification of individual RNA species.
(B) Diagram showing the use of TCEP to extinguish fluorescence signals via cleavage of disulfide bonds that link the fluorescent dye to the readout probe. For more details on the developed protocols review Chen et al. 2015 and Moffitt et al. 2016.
Design of readout probes
When designing readout probes, several important considerations need to be taken into account.
|
1.
|
To improve binding efficiency, select probes that have similar Tm values and GC content so that their hybridization properties are similar.
|
|
2.
|
Limit the number of potential off-target binding sites. Screen sequences for homology to RNAs in the transcriptome studied.
|
|
3.
|
Sequences must be orthogonal in that they should have limited homology with one another to prevent binding of one readout probe to the wrong readout sequence.
|
The following steps are required for the design of new or additional readout probes:
|
Step 1
|
- Utilize existing sets of orthogonal nucleic acid sequences.
- Make sure that readout probes have little homology to other readout probes to prevent off-target binding.
- Readout sequences of 30-nt length appear to work best. These can be created from25-nt sequences by concatenating portions of the probes or by adding five random nucleotides to either end (see http://elledgelab.med.harvard.edu/?page_id=638).
|
|
Step 2
|
- Remove potential probes with homology to members of the targeted transcriptome. Create a BLAST library to the transcriptome and BLAST each potential readout probe sequence against this library.
- Remove any probe that contains a contiguous stretch of homology longer than fourteen (14) nt.
|
|
Step 3
|
- Remove potential readout probes that contain significant homology to one another.
- Select a subset of possible readout probes and build a BLAST database for these sequences and use BLAST for the identification of homologous regions.
- Exclude probes with homologous regions longer than ten (10) nt.
|
|
Step 4
|
- Synthesize or order these probes (from Bio-Synthesis Inc.). Probes are usually tagged on the 3’ end with a Cy5 fluorophore. Use HPLC purified probes at the 100 nmol scale.
|
Readout probe sequences
|
Bit
|
Readout probe
|
Sequence
|
Dye
|
|
1
|
RS0015
|
ATCCTCCTTCAATACATCCC
|
Cy5
|
|
2
|
RS0083
|
ACACTACCACCATTTCCTAT
|
Alexa750
|
|
3
|
RS0095
|
ACTCCACTACTACTCACTCT
|
Alexa750
|
|
4
|
RS0109
|
ACCCTCTAACTTCCATCACA
|
Cy5
|
|
5
|
RS0175
|
ACCACAACCCATTCCTTTCA
|
Cy5
|
|
6
|
RS0237
|
ACCCTTTACAAACACACCCT
|
Alexa750
|
|
7
|
RS0247
|
TTTCTACCACTAATCAACCC
|
Cy5
|
|
8
|
RS0255
|
TCCTATTCTCAACCTAACCT
|
Alexa750
|
|
9
|
RS0307
|
TATCCTTCAATCCCTCCACA
|
Alexa750
|
|
10
|
RS0332
|
ACATTACACCTCATTCTCCC
|
Cy5
|
|
11
|
RS0343
|
TTTACTCCCTACACCTCCAA
|
Cy5
|
|
12
|
RS0384
|
TTCTCCCTCTATCAACTCTA
|
Alexa750
|
|
13
|
RS0406
|
ACCCTTACTACTACATCATC
|
Cy5
|
|
14
|
RS0451
|
TCCTAACAACCAACTACTCC
|
Alexa750
|
|
15
|
RS0468
|
TCTATCATTACCCTCCTCCT
|
Alexa750
|
|
16
|
RS0548
|
TATTCACCTTACAAACCCTC
|
Cy5
|
Note: The dye is attached to the probe via a disulfide bond at the 5’end.
Reference
Filonov & Jaffrey (ed.); Visualizing RNA Dynamics in the Cell. Methods in Enzymology, Volume 572 (2016).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S., & Zhuang, X. (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science (New York, N.Y.), 348(6233), aaa6090. http://doi.org/10.1126/science.aaa6090.
Jeffrey R. Moffitt, Junjie Hao, Guiping Wang, Kok Hao Chen, Hazen P. Babcock, and Xiaowei Zhuang; High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. PNAS 2016 : 1612826113v1-201612826.
Ordering & Contact Information
- Please contact us for additional information or send an email to info@biosyn.com.
- You may also request an online quote.
- To contact us by phone, please call 1-972-420-8505 or Fax at 1-972-420-0442
- Orders may be placed using a purchase order (PO) or by credit card through our secure online ordering system.
- When using a credit card (
 ) it will be billed under "Bio-Synthesis, Inc."
) it will be billed under "Bio-Synthesis, Inc."
-.-