Solid Phase Peptide Synthesis (SPPS)
The control and regulatory mechanisms for many biological processes are dependent on peptides and proteins derived from α–amino acids. In addition, many modern medicines are now produced from peptides or derivatives of peptides. A few examples are anti-cancer agents, antibiotics or peptide based drugs that control blood pressure. For this reasons α–amino acids and peptide chemistry has become a central technology in organic chemistry, biochemistry, biotechnology and medicinal chemistry. Solid phase peptide synthesis has become the major automated synthesis method or technology used for the production of synthetic peptides.
The automation of solid phase peptide synthesis and the many improvements made in peptide synthesis instrumentation in recent years have made synthetic peptides and their derivatives more available to the scientific community and the biological industry as a whole. Synthetic peptides can be synthesized automatically using solid phase peptide synthesis and used to manufacture epitope-specific antibodies, map antibody epitopes and study enzyme binding sites or to design and synthesize novel peptide- or protein-mimetics or even whole enzymes.
To synthesize a peptide bond between two amino acids will need to be formed. The exact size of peptides is not well defined but it usually refers to flexible chains of up to 30 to 50 amino acids. However, peptides with up to 100 amino acids within the chain can now be synthesized.
The basic concept in solid phase peptide synthesis is the step-wise construction of a peptide chain attached to an insoluble polymeric support (see Figure 1. for general synthesis scheme).

Figure 1: General Scheme for Solid Phase Peptide Synthesis
This approach permits unreacted reagents to be removed by washing without loss of product. Synthesis of the peptide chain proceeds from the carboxyl end of the amino terminus of the peptide.
The carboxyl moiety of each incoming amino acid is activated by one of several strategies and couples with the α-amino group of the preceding amino acid. The α-amino group of the incoming residue is temporarily blocked in order to prohibit peptide bond formation at this site. The residue is de-blocked at the beginning of the next synthesis cycle. In addition, reactive side chains on the amino acids are modified with appropriate protecting groups. The peptide chain is extended by reiteration of the synthesis cycle. Excess reagents are used to drive reactions as close to completion as possible. This generates the maximum possible yield of the final product.
After fully assembling the peptide the side-chain protecting groups are removed, and the peptide is cleaved from the solid support, using conditions that inflict minimal damage on labile residues. The product is analyzed to verify the sequence thereafter. The synthetic peptide is usually purified by gel chromatography or HPLC. The blocking group used for blocking the α-amino group determines both the synthesis chemistry employed and the nature of the side-chain protecting groups. The two most commonly used α-amino protecting groups are Fmoc (9-fluorenyl-methoxy-carbonyl) and tBoc (tert.-butyloxycarbonyl). Fmoc side-chain protection is provided by ester, ether and urethane derivatives of tert.-butanol, while the corresponding tBoc protecting groups are ester, ether, and urethane derivatives of benzyl alcohol. The latter are usually modified by the introduction of electron-withdrawing halogens for greater acid-stability. Ether and ester derivatives of cyclopentyl or cyclohexyl alcohol are also employed.
The Fmoc protecting group is base-labile. It is usually removed with a dilute base such as piperidine. The side-chain protecting groups are removed by treatment with trifluoroacetic acid (TFA), which also cleaves the bond anchoring the peptide to the support. The tBoc protecting group is removed with a mild acid (usually dilute TFA). Hydrofluoric acid (HF) can be used both to deprotect the amino acid side chains and to cleave the peptide from the resin support. Fmoc is a gentler method than tBoc since the peptide chain is not subjected to acid at each cycle and has become the major method employed in commercial automated peptide synthesis.
Fmoc Chemistry
The α-amino groups are protected by the Fmoc (9-fluorenylmethoxycarbonyl) group, while side-chain protection is provided by ester, ether and urethane derivatives of tert.-butanol (see Figure 2. for details).
.jpg)
Figure 2. Fmoc-Chemistry for Peptide Synthesis
Carboxyl group activation of the incoming amino acid utilizes one of the following methods:
1. BOP/HOBt/NMM:
The amino acid is mixed with the BOP (Castro's reagent or (benzotriazol-yloxy) tris (dimethylamino) phosphonium hexa-fluoro-phosphate), HOBt (1-hydro-xybenzotriazole) and sufficient NMM (N-methyl-morpholine) to ionize 50% of the HOBt.
2. DIPCDI:
DIPCDI (disopropylcarbo-diimide) can be used either with or without HOBt in a solution of DCM:DMF (di-chloromethane and dimethyl-formamide).
3. Active esters:
These are usually pentafluorophenyl esters (OPfp) of the amino acids. The pentafluorophenyl esters of serine and threonine are oils and therefore are employed in the more convenient form of the dihydrooxobenzotriazine ester (ODhbt).
tBoc Chemistry
tBoc (tert-butyl-oxy-carbonyl) chemistry is widely used in peptide synthesis (see Figure 3.).
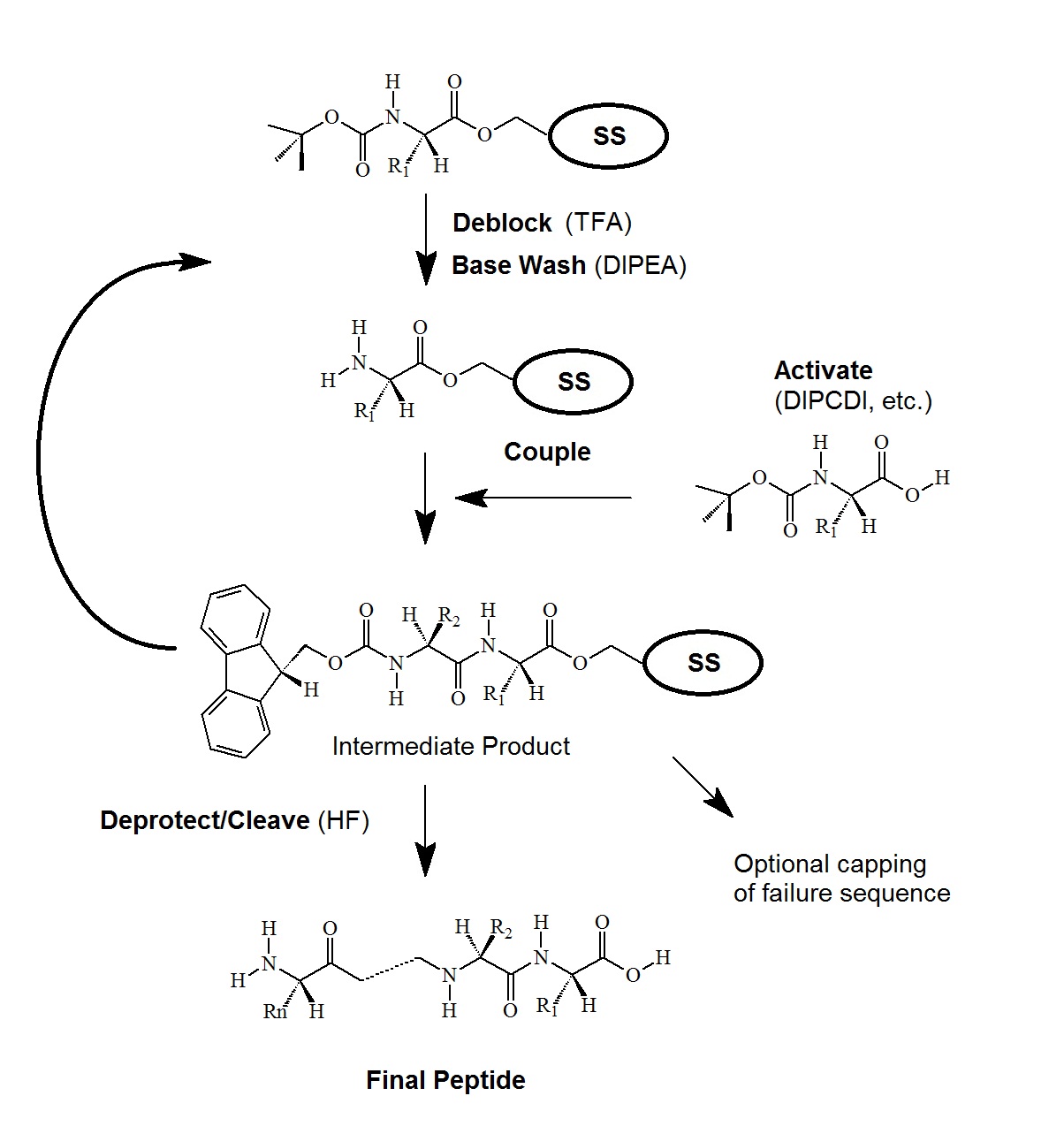
Figure 3. tBoc-Chemistry for Peptide Synthesis
Side chain protection is done by ester, ether, and urethane derivatives of benzyl alcohol, and modified by the introduction of electron-withdrawing halogens for greater acid-stability. Ether and ester derivatives of cyclopentyl or cyclohexyl alcohol are also employed.
Activation of the incoming amino acid employs DPCDI in a DCM:DMF solution and may also include HOBt.
Peptide Synthesis Chemistries: A Comparison
Fmoc-Protected L-Amino Acid Derivatives
Fmoc amino acids use usually t-butyl based protection for most labile side-chains. The trityl (Trt) protecting group is recommended for Cys. This is removed when the peptide is cleaved with TFA. The acetamidomethyl (Acm) derivative of Cys is unaffected by TFA treatment, but may be rapidly removed by electrophilic reagents such as mercury or iodine. Oxidative deprotection with iodine yields disulfides directly and is a convenient, clean method of forming intramolecular disulfide links. For Arg side-chain protection, both Mtr and the more easily removed Pmc group are widely used. The amidated side-chains of Gln and Asn are protected either with the standard Tmob or with the more recently available Trt.
Fmoc-Protected D-Amino Acid Derivatives
Peptides containing D-amino acids are finding increased use in both research laboratory and pharmaceutical application. Their presence in the peptide allows chain configuration not possible with the L-amino acids alone. Many potent, naturally occurring antibiotics and toxins are cyclic peptides, and often incorporate several D-amino acids to confer a specific conformation as well as make the peptide more resistant to proteolytic attack.