Peptide Mimetics
Modern day medicinal chemistry research is relying more and more on biotechnology. Now, molecular biology research has become a driving force behind the screening and discovery of macromolecular targets. Biotechnological companies usually focus on peptide and protein therapeutics, although these molecules have intrinsic associated problems such as low bioavailability, rapid metabolism, and lack of oral activity. To circumvent these problems, researchers may rely upon chemical synthesis to generate non-peptide molecules for drug discovery. The challenge is to design strategies that combine the strengths of biotechnology to discover molecular targets and medicinal chemistry to develop pharmaceutically useful compounds.
The terms “peptide mimetics” and “peptidomimetics” have been used interchangeably for the description of compounds discovered through a variety of research strategies, including compounds identified by random screening approaches. A peptide mimetic can be a molecule such as a peptide, a modified peptide or any other molecule that biologically mimics active ligands of hormones, cytokines, enzyme substrates, viruses or other bio-molecules. This peptide mimetic may antagonize, stimulate, or otherwise modulate the physiological activity of the natural ligands. Peptides that have a length of 14 to 20 amino acids can serve as lead compounds for the development of therapeutically effective agents. Since they are usually considerably smaller than the parent molecule, chances are high that they may show less side effects and are easier to deliver to the target. The major emphasis for synthesizing a peptide mimetic is to mimic a peptide or protein ligand of receptors. However the technology is useful for enzyme inhibition as well. Since the term “peptide mimetic” is relatively broad, natural compounds such as natural occurring opiate alkaloids are examples of peptidomimetics as well. Indeed, small peptides, as well as opiods, can sometimes target the same receptor. An example is zyklophin, a cyclic peptide that is a selective peptide kappa opioid receptor antagonist, showing biological activity that may have a potential to be a lead compound for the development of potential therapeutic agents. Furthermore, nonpeptides that are structurally different from their peptide parents (lacking flexibility, amide bonds, and obvious pharmacophore similarity), can lead to highly selective ligands for subtypes of receptors in both peptide and nonpeptide compounds. A pharmacophore is a compound with the molecular features that are necessary for molecular recognition of a ligand by a biological macromolecule. Peptide mimetics can be discovered by screening through compound libraries or designed as de novo mimetics. In this case, the design is centered on the replacement of individual peptide bonds by a nonpeptidic moiety or other spacer group. Unnatural amino acids, isosteric replacements, cyclic peptide derivatives, bond surrogates, spacers or parts of molecules with conformational constraints, as well as larger subunits, for example, dipeptide mimetics, can be incorporated into the peptides. These compounds bridge the gap between simple peptide analogs and completely nonpeptide structures. Information gained from the study of structure-activity relationships and conformational properties of the peptide structures is needed to make a rational design feasible. General principles on how to design peptide mimetics have been established during the last 20 years (Olsen et al 1993; Farmer 1980).
Criteria for the Design of Peptide Mimetics
- Replace as much of the backbone as possible by a nonpeptide framework. A structural template may need to be designed to eliminate amide bonds.
- Maintain peptide side chain pharmacophoric groups as they are found in the peptide. As an example, if a lysine residue is known to be required for activity in the peptide, then the first generation mimetic should have a primary amino group at the end of a methylene chain. During the progression of the design different chain lengths, conformational constraints, and substitutions on the nitrogen may generate enhanced binding affinities.
- Conformational flexibility should be retained in the first generation mimetics. The first round of design should leave some of the side chain pharmacophoric groups unconstrained so that they can adopt conformations of peptide analogs. If a lead activity is seen for a flexible mimetic, adding conformational constrains to side chain groups may be the approach to enhance potency and selectivity.
- Select targets based on availability of a pharmacophore hypothesis. Alternatively, develop the information as a first step. If no information of structure-activity relationships and no three-dimensional hypothesis of the bioactive compound is available it is irrational to design a mimetic. However, lead compounds from screening experiments may be useful compounds to start the design of a peptide mimetic.
A natural or common approach is to mimic the architectural elements of peptides and protein structures such as ß-turns, helices and ß-sheets. Major efforts have been devoted to develop templates that mimic or stabilize these common architectural elements. Figure 1 illustrates the classical protein structures of helices and turns present in 3D structures of proteins.
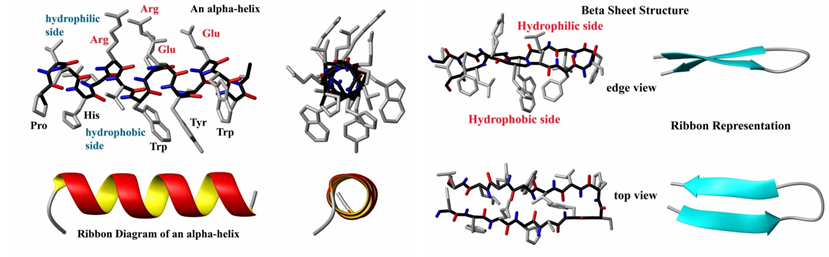
Figure 1: A: Structure and ribbon diagram of a protein alpha helix (left panel). The backbone atoms form a coil (black bonds) while the carbonyl groups (red) form hydrogen bonds with the amide groups (blue). B: Structure and ribbon diagram of a protein beta sheet (left panel). Note the oscillating positions of the carboxyl (red) and amide (blue) groups which form hydrogen bonds between the two beta strands (source: http://en.citizendium.org/wiki/Protein_structure).
Figure 2 illustrates concepts in the rational design of biological active peptides and peptidomimetics.
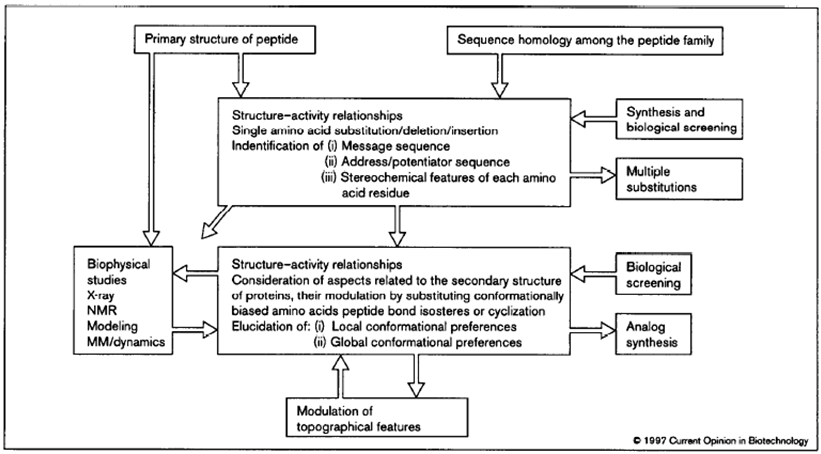
Figure 2: Flowchart highlighting the iterative approach to the rational design process of peptidomimetics. The primary structure of a lead peptide may be identified, for example, using natural product screening, phage display, synthetic libraries or using anti-idiotypic antibodies. Multiple substitutions of amino acids can be generated via analog synthesis or via mutagenesis after making a library based upon the lead peptide. (Source: Kieber-Emmons T, Murali R, Greene MI. Therapeutic peptides and peptidomimetics. Curr Opin Biotechnol. 1997 Aug;8(4):435-41).
ß-turn Mimetics
The majority of the ß-turn mimetics have been dipeptide mimetic replacements for the i + 1 and i + 2 residues at the corner of the turn. The mimetics are usually designed such that they retain the intramolecular hydrogen bonding network of the neighboring peptide chain. Since the side chains in the peptide turns are mostly exposed at the surface of a protein and therefore are most likely be involved in intramolecular interactions with a target protein, such as a receptor, the ß-turn mimetics may need to be designed to closely resample the original ß-turn structure to retain or achieve better binding activity. The 9-membered ring lactam system illustrated in Figure 3 (left panel, upper corner) was designed to replace the interchain hydrogen bond between residues i and i + 3 with a methylene group and to replace all of the amide bonds except for the exposed corner with carbon-carbon bonds (Olsen et al., 1993). Many groups have designed and characterized similar specific target peptide turns. A diversity of structures have been used to design inhibitors for glycoprotein IIb/IIIa (GpIIbIIIa) binding to fibrinogen receptors. Glycoprotein IIb/IIIa (gpIIb/IIIa, also known as integrin αIIbβ3) is an integrin complex found on platelets. It binds to fibrinogen and aids in platelet activation. The complex is formed via calcium-dependent association of gpIIb and gpIIIa during normal platelet aggregation and endothelial adherence.
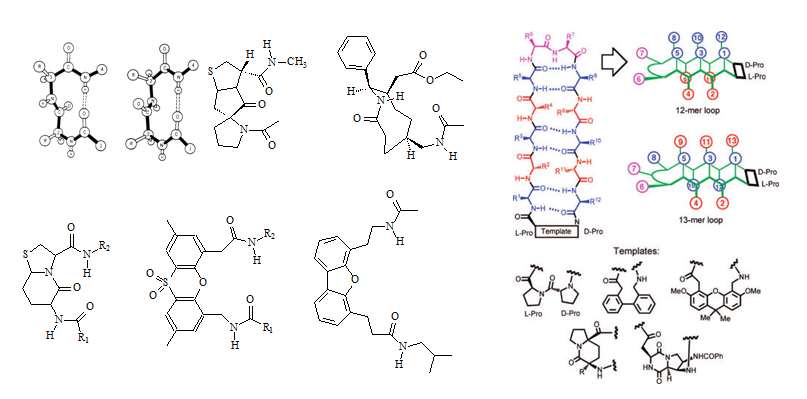
Figure 3: Structures of ß-turns, ß-turn templates for mimetics as well as a prototypical template bound 12-mer ß-hairpin loop are illustrated in this figure (Source: Olson et al., 1993; Robinson 2008). Two types of ß-turns (Type-I and Type-II) are depicted in the upper left of the left panel (Source: http://imtech.res.in/raghava/betatpred/intro.html). The inhibitory RGD(S) peptide sequence has been used as a lead sequence. Peptidomimetics have been made by constricting the sequence into a cyclic peptide and several nonpeptide mimetics have been described as well. Benzene rings, steroids, and benzodiazepines have been used as templates.
a-Helix Mimetics
The α-helix is one of the best characterized features found in protein structures. Compared to the β-turn, the α-helix incorporates more amino acid residues, 3.4 residues per turn. If the recognition of a larger helix involves many residues, the design of a series of mimetic changes would be impractical because the required templates would be too complex, since a large number of side chain mimetics would be needed.
Two cases of α-helices may allow for a successful design of mimetics:
A: The residues involved in biological recognition lie along one face of the helix. This allows the design of a small bridging template similar to a β-turn template.
B: The helix performs a function as a spacer and no individual amino acid is required for activity within the helix segment
An example for case B is the study of vasoactive intestinal peptide (VIP) by Bolin’s group where an “alanine scan” of the peptide sequence was performed by synthesizing alanine analogs of the VIP peptide (Donnel et al., 1991). Each individual residue within the sequence was replaced with an alanine. Together with NMR studies by Fry et al. 1989, it was shown that the peptide in solution was highly helical. These results suggested that the sequence region between Tyr 10 and Tyr 22 performs as a helical spacer, having no direct side-chain interaction with the VIP receptor. Results like this suggest that properly designed mimetics have the potential to replace the polypeptide spacer helix. However, issues as solubility and potency of the hybrids will have to be addressed.
|
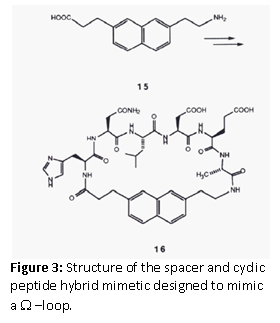
Ω Loop Mimetics
Leszczynski et al. (1986) designed a spacer to constrain the ends of a loop sequence. Sarabu et al. (1993) used the concept to design a Ω –loop mimetic for a prominent loop structure in the immunomodulatory cytokine, IL-1α. The omega loop is a protein motif that consists of a loop of any length and any amino acid sequence with the requirement that residues that make up the beginning and end of the loop are very close together. It is named after its shape, which resembles the Greek capital letter Omega. The spacer 7-(2-aminoethyl)napthtalene-2-propionic acid, was chosen to design a cyclic peptide hybrid, which was used for binding assays. However, since no binding was detected it was concluded that this loop may not be involved in receptor binding.
|
 |
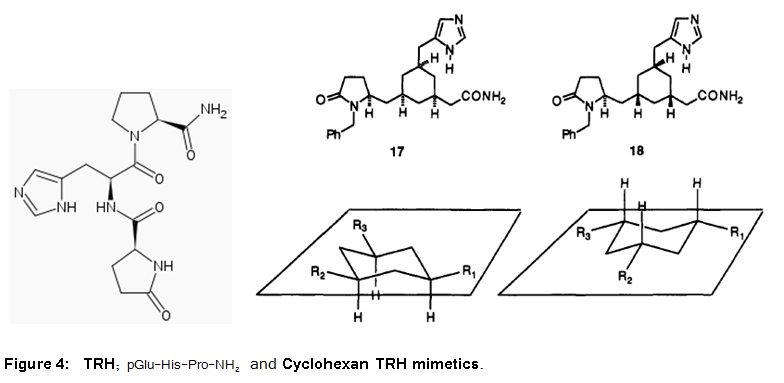
Mimetics Based on the Cyclohexane Framework
The thyrotropin-releasing hormone (TRH), also called thyrotropin-releasing factor (TRF), thyroliberin or protirelin, is a peptide hormone that stimulates the release of thyroid-stimulating hormone (TSH) and prolactin from the anterior pituitary. It has been extensively studied in the past and crystal and solution structures are available. Furthermore, models have been proposed for bioactive conformations (Olson et al., 1993). The 1,3,5-cis-trisubtituted cyclohexane framework was chosen for the design of a mimetic (Figure 4). This peptidomimetics represent efforts to translate peptide and protein structures into small molecular weight, nonpeptide compounds that truly mimic their biological function.
Protein Domain Mimics
Recently Ni et al. (2010) have identified a fragment antigen-binding (Fab) 1D05 which binds PCSK9 with nanomolar affinity. They showed that the fully human antibody 1D05-IgG2 completely blocks the inhibitory effects of wild-type PCSK9 and two gain-of-function human PCSK9 mutants, S127R and D374Y, respectively. The crystal structure of 1D05-Fab bound to PCSK9 revealed that 1D05-Fab binds to an epitope on the PCSK9 catalytic domain which includes the entire LDL receptor (LDLr) EGF(A) binding site. Notably, the 1D05-Fab CDR-H3 and CDR-H2 loops structurally mimic the EGF(A) domain of LDLr. This was revealed by the crystal structure of the PCSK9/1D05-Fab complex. The research group could show that 1D05 acts as a structural mimic of the EGF(A) domain of LDLr and sterically prevents PCSK9 from binding to the receptor. Proprotein convertase subtilisin-like/kexin type 9 (PCSK9) regulates LDL cholesterol levels by inhibiting LDLr-mediated cellular LDL uptake.
Stabled Peptides
|
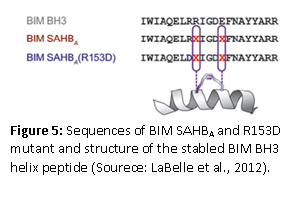
Synthetic peptides that specifically bind to proteins such as cytokines, protein hormones, and nuclear hormone receptors offer an alternative approach to small molecules for the modulation of protein and receptor signaling and subsequent gene expression. LaBelle et al (2012) published a research article that showed how a stable helical BCL-2-interacting mediator of cell death (BIM) peptide overcomes apoptotic resistance in hematologic cancers. A hydrocarbon-stabled peptide modeled after the BIM BH3 helix broadly targeted BCL-2 family proteins with high affinity. This peptide blocked inhibitory antiapoptotic interactions, thereby directly triggering proapoptotic activity, inducing dose-responsive and BH3 sequence-specific cell death of hematologic cancer cells. The therapeutic potential of the peptide was established by the selective activation of cell death in the aberrant lymphoid infiltrates of mice reconstituted with BIM-deficient bone marrow and in a human AML xenograft model. These results indicate that the BCL-2 family interaction network that lies at the crossroads of the cell’s life-death decisions can be targeted with therapeutic compounds that selectively modulate the network. Apoptosis regulator Bcl-2, BH is a family of evolutionarily related proteins that govern mitochondrial outer membrane permeabilization (MOMP) and can be either pro-apoptotic or anti-apoptotic. There are a total of 25 genes in the Bcl-2 family known to date.
|
 |
Stapled peptides may offer new ways to treat so called “undruggable” diseases. Available drugs, primarily small molecules and therapeutic proteins, address only an estimated 10% to 20% of all identified therapeutic targets within the human body. What makes stabled peptides different? Proteases, naturally present in the human body, can only recognize and digest peptides when they are unfolded. Therefore, if the peptides are locked into certain folded shapes, they are protected from the attack of the proteases and will remain longer in the tissues where the targeted compounds reside. In the past, many attempts have been made to stabilize peptides by chemical modifications, however, often some if not all of their biological activity is lost or the modified peptides cannot penetrate cells. Key steps for the design of stabling peptides involves using a cross-linking chemistry that locks the peptides into an α-helical shape that mimics the structure found at the interface of many protein-protein interactions. One prominent method is the hydrocarbon stapling approach. What makes the peptide cell permeable? The following researchers think they have the answer. Muppidi et al., in 2012, showed that the direct chemical modifications of helical peptides have provided a simple and effective means to ‘translate’ bioactive helical peptides into potential therapeutics targeting intracellular protein-protein interactions. The researchers have shown that the distance-matching bisaryl cross-linkers can reinforce peptide helices containing two cysteines at the i.i+7 positions and confer cell permeability to the cross-linked peptides. The group reported the first crystal structure of a biphenyl cross-linked Noxa peptide in complex with its target Mcl-1 at a 2.0 Å resolution. Guided by the structure, they remodeled the surface of the cross-linked peptide through side chain substitution and N-methylation, and obtained a pair of cross-linked peptides with substantially increased helicity, cell permeability, proteolytic stability, and cell-killing activity in Mcl-1-overexpressing U937 cells. Dimartino’s group described the solid-phase synthesis of hydrogen-bonded surrogate-derived artificial α-helices by a ring-closing metathesis reaction. The researchers found that the Hoveyda-Grubbs catalyst gave high yields of the macrocycle irrespective of the peptide sequence. Furthermore, they concluded that hydrogen-bonded surrogate derived artificial α-helices can be efficiently synthesized on solid phases with this catalyst and that the synthesis of the α-helices does not require the preparation of new enantiomerically pure amino acids. Also they can be achieved by standard Peptides Synthesis protocols with easily available monomers. The number of researchers that now report on the synthesis of these peptides has increased considerably since 1998 when Blackwell and Grubbs in 1998 reported the synthesis of the first generation of stapled peptides ranging in size from 12 to 35 amino acids in length.
Harrison et al., published a paper in 2010 that reported the downsizing of proteins to short water-stable α-helices that maintain biological potency. The much shorter cyclic peptides mimic the α-helical parts of the protein structure appear to be more stable than the parent proteins, and have biological activity as well. The building block is a small cyclic pentapeptide (KAAAD), which structure is similar to a lactam between residues 1 and 5. The joining of two or more of these peptides allows the design of stable peptides with more turns, whereas the replacement of alanine allows for the synthesis of more possible mimics of the endogenous protein. The researchers showed that an analog of nociceptin is a potent agonist at ORL-1 (Figure 6). Furthermore, the constrained peptides are stable in human serum for over 24 hours, as compared to their uncyclized peptides, which are degraded rapidly.
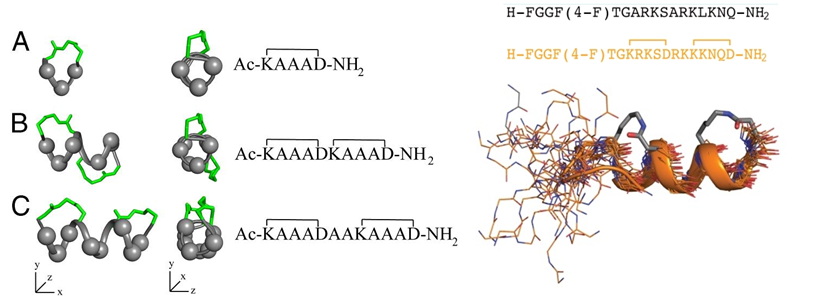
Figure 6: (Left) α-helices composed of cyclic pentapeptides. Different distributions of variable side chains (gray spheres) and linking bridges (green) allow the design of different helix mimics for binding to different receptors. (Right) GPCR-ligand nociceptin. The nociceptin peptide with residues 1 to 17 is a super agonist of ORL-1. ORL 1 or Opioid Like Receptor 1 is a member of the family of rhodopsin-like G protein-coupled receptors (GPCR) that shares functional and structural homology with Opioid Receptors systems and is involved in a variety of biomedical important processes, such as anxiety, nociception, feeding, and memory. The mimic of this peptide is shown below the sequences (Harrison et al. 2010).
Verdine and Hilinski in 2010 report on the design and synthesis of a new class of synthetic miniproteins locked into their bioactive α-helical fold through the site-specific introduction of a chemical brace, an all-hydrocarbon stable in ‘Methods of Enzymology.’ The researchers discuss considerations crucial to the successful design and evaluation of potent, stapled peptide interactions to facilitate the broad application of this technology to intractable targets of both basic biologic interest and potential therapeutic value. The synthesis of three types of all-hydrocarbon stapled peptides is described (Figure 7). The synthesis of stapled peptides is usually performed with standard Fmoc-chemistry based solid phase synthesis with modifications to allow for the cyclization reaction by metathesis prior to labeling or tagging the peptide with fluorophores, biotin or other functional groups (See Verdine and Hilinski, 2012, for more details). Extensive biophysical characterization can be done with a diverse portfolio of techniques such as Circular Dichroism (CD), to evaluate the presence and nature of the α-helical structure, Fluorescence Polarization (FP), and Surface Plasmon Resonance (SPR), to quantitatively measure the binding constants that describe the interaction of stabled peptides with a recombinant target protein. Furthermore, the presence of the final peptide as well as proteolytic susceptibility can be measured using assays based on High-Performance Liquid Chromatography (HPLC)- or Mass Spectrometry (MS)-based detection of intact peptides. The cell permeability of the peptides can be evaluated using flow cytometry and confocal fluorescence microscopy, which require fluorescently labeled peptides. The ability of the peptides to interact with the target can be tested in vitro by in vitro immunoprecipitation (IP) and pull-down assays. The final step toward validating the therapeutic usefulness of the finally selected peptide mimetic is a test for in vivo efficacy. A relevant biological model system, such as a tumor progression murine model for cancer, is needed for this test. This is a final proof-of-principle in vivo experiment before clinical trials can be started.
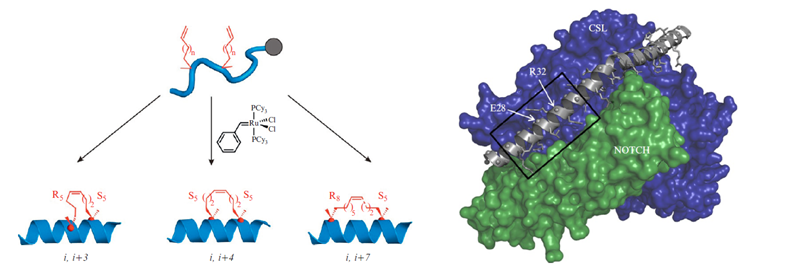
Figure 7: (Left) α-Methyl, α-alkenylglycine cross-linking amino acids are incorporated during solid-phase peptide synthesis. The schematics for the synthesis of an i, i+3 stapled, an i, i+4 stapled, and an i, i+7 stapled peptide is illustrated. (Right) The 3D model of a stapled peptide targeting the NOTCH/CSL binary complex is shown. A kinked α-helix from the MAML transcriptional coactivator protein binds to a cleft created by the NOTCH/CSL complex. The region of MAML from which the bioactive SAHM1 stapled peptide was designed is indicated with a box. MAML residues E28 and R32 were replaced with S5 cross-linking amino acids to form SAHM1. PDB: 2F8X. (Source: Verdine and Hilinski, 2012).
To conclude, rational drug design has benefited from technological progress made in recent years. Advances in laboratory processes, particularly in mass spectrometry instrumentation and techniques, nuclear magnetic resonance technology (NMR), associated synthetic schemes, genomics, computational chemistry, computer software and power, and screening technology have contributed to an increase of rational drug design. Peptidomimetics and peptides have the potential to become novel pharmaceuticals that can affect unique aspects of receptor assembly or ligand development that cannot be achieved by conventional pharmaceuticals. Important applications include the use of retro-inverso peptides to induce long lasting immune responses and to target T cell receptors. Briefly, D-retro-inverso-peptide share similar arrangements of side-chains. However, their carboxyl and amino groups point in opposing directions. For small peptides it is thought that an L-peptide and its D-retro-inverso-peptide will most likely have a similar binding affinity with its target L-protein.
References
Jane V. Aldricha,1, Kshitij A. Patkara, and Jay P. McLaughlinb,c; Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short. duration of action. PNAS October 27, 2009 vol. 106 no. 43 18396-18401.
Helen E. Blackwell, Robert H. Grubbs Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis; Angewandte Chemie International Edition Volume 37, Issue 23, pages 3281–3284, December 17, 1998.
Gianluca Dimartino, Deyun Wang, Ross N. Chapman, and Paramjit S. Arora; Solid-Phase Synthesis of Hydrogen-Bond Surrogate-Derived a-Helices ORGANIC LETTERS 2005 Vol. 7, No. 12 2389-2392
Fry, D. C.; Madwon, V. S.; Bolii, D. R.; Greeley, D. N.; Toome, V.; Wegrzynski, B. B. Solution Structure of an Analog of Vasoactive Intestinal Peptide as Determined by Two-Dimensional NMR and Circular Dichroism Spectroecopies and Constrained Molecular Dynamics. Biochemistry 1989,2899-2409.
Rosemary S. Harrison, Nicholas E. Shepherd, Huy N. Hoang, Gloria Ruiz-Gómez, Timothy A. Hill, Russell W. Driver, Vishal S. Desai, Paul R. Young, Giovanni Abbenante, and David P. Fairlie; Downsizing human, bacterial, and viral proteins to short water-stable alpha helices that maintain biological potency. Proc Natl Acad Sci U S A. 2010 June 29; 107(26): 11686–11691
Kieber-Emmons T, Murali R, Greene MI. Therapeutic peptides and peptidomimetics. Curr Opin Biotechnol. 1997 Aug;8(4):435-41.
James L. LaBelle, Samuel G. Katz, Gregory H. Bird, Evripidis Gavathiotis, Michelle L. Stewart, Chelsea Lawrence, Jill K. Fisher, Marina Godes, Kenneth Pitter, Andrew L. Kung, and Loren D. Walensky; A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012 June 1; 122(6): 2018–2031.
Leszczynski, J. F.; Rose, G. D. Loops in Globular Proteins: ANovel Category of Secondary Structure. Science 1986,234, 849-855.
Avinash Muppidi , Kenichiro Doi, Selvakumar Edwardraja, Eric J Drake, Andrew M. Gulick, Hong-Gang Wang , and Qing Lin; Rational Design of Proteolytically Stable, Cell-Permeable Peptide-Based Selective Mcl-1 Inhibitors J. Am. Chem. Soc., Just Accepted Manuscript. 2012.
Yan G. Ni , Stefania Di Marco , Jon H. Condra , Laurence B. Peterson , Weirong Wang , Fubao Wang , Shilpa Pandit , Holly A. Hammond , Ray Rosa , Richard T. Cummings , Dana D. Wood , Xiaomei Liu , Matthew J. Bottomley , Xun Shen , Rose M. Cubbon , Sheng-ping Wang , Douglas G. Johns , Cinzia Volpari , Lora Hamuro , Jayne Chin , Lingyi Huang , Jing Zhang Zhao , Salvatore Vitelli , Peter Haytko , Douglas Wisniewski , Lyndon J. Mitnaul , Carl P. Sparrow , Brian Hubbard , Andrea Carfí , and Ayesha Sitlani; A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. The Journal of Lipid Research, 52, 78-86 (2010).
O'Donnell, M.; Garippa, R. J.; O'Neill, N. C.; Bolin, D. R.; Cotrell, J. M. Structure-Activity Studies of Vaeoactive Intestinal Polypeptide. J. Biol. Chem. 1991, 266,63894392.
Gary L. Olson,. David R. Bolin, Mary Pat Bonner, Michael BOs, Charles M. Cook, David C. Fry, Bradford J. Graves, Marcos Hatada, David E. Hill, Michael Kahn, Vincent S. Madison, Victoria K. Rusiecki, Ramakanth Sarabu, Jerry Sepinwall, George P. Vincent, and Matthew E. Perspective: Concepts and Progress in the Development of Peptide Mimetics. JOURNAL OF MEDICINAL CH EM ISTRY Volume 36, Number 21 October 15,1993.
JOHN A. ROBINSON ⌈-Hairpin Peptidomimetics: Design, Structures and Biological Activities. ACCOUNTS OF CHEMICAL RESEARCH 1278-1288 October 2008 Vol. 41, No. 10.
Sarabu, R.; Lovey, K.; Madison, V. S.; Fry, D. C.; Greeley, D. N.; Cook,C. M.; Oleon, G.L. Design, Synthssis and Three Dimensional Structural Characterization of a Constrained Ω -loop Excised from Interleukin-la. Tetrahedron 1993,49,3629-3640.
Verdine GL, Hilinski GJ.; Stapled peptides for intracellular drug targets. Methods Enzymol. 2012; 503:3-33.