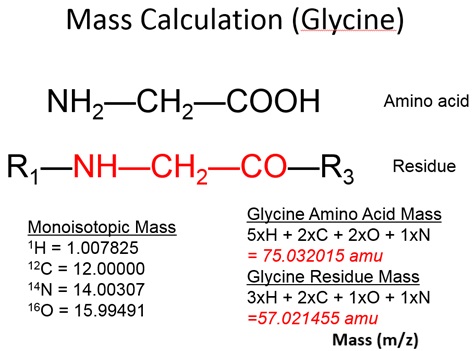For a general object, if the density and the volume of that object are known, the mass of that object can be derived using the following equation:
m =ρ x V;
where m= mass, ρ = density, and V = volume.
See also: http://hyperphysics.phy-astr.gsu.edu/hbase/mass.html, https://www.calculator.net/mass-calculator.html.
However, in a mass spectrometer, ions are acquired as m/z values. In matrix-assisted laser desorption time of flight mass spectrometer instruments, the major ions for peptides analyzed are usually parent ions for which z =1. The shape of the mass or ion peak determines the resolution. Based on the shape of the peak the resolution can be mathematically described using the formula:
Resolution = M/ΔM
where M = mass and ΔM (dMass or delta Mass) = mass range at 50% intensity of the peak.
The resolving power in a mass spectrometer is defined as the ability of the instrument or measurement to distinguish between two peaks at m/z values differing by a small amount, expressed as the peak width in mass units. See also: https://en.wikipedia.org/wiki/Full_width_at_half_maximum.
Example 1: Resolving Power and Accuracy
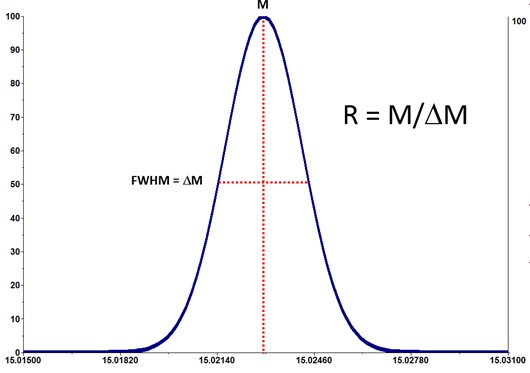
Legend: M = mass; FWHM = ΔM = full width at half maximum; ΔM = delta Mass.
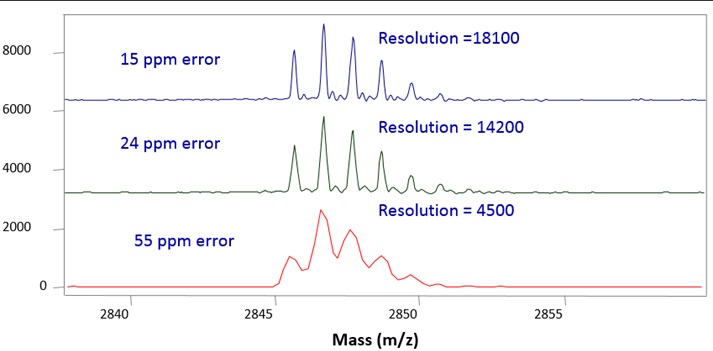
However, the accuracy of mass measurements depends on its resolution. Therefore, a higher resolution results in a better mass accuracy.
Example 2: Impact of Resolution and Error of Measurement
Calculating Peptide Masses
• Sum the monoisotopic residue masses
• Add mass of H2O (18.01056)
• Add mass of H+ (1.00785 to get M+H)
• If Met is oxidized add 15.99491
• If Cys has acrylamide adduct add 71.0371
• If Cys is iodoacetylated add 58.0071
• Other modifications are listed at
– http://prowl.rockefeller.edu/aainfo/deltamassv2.html
• Mostly consider peptides with masses > 400 for proteomic experiments
Example 3: Mass Calculation for a Glycine Ion