Antisense oligonucleotides slow the progression of the pediatric neurodegenerative disorder Batten’s disease by correcting mRNA splicing, paving the way for personalized medicine
Neuronal ceroid lipofuscinoses (NCLs) refer to a group of neurodegenerative disorders, which negatively impacts vision, behavior, learning, speech, etc., causing seizure, impaired motor skill, reduced intellectual capacity and early death. The NCLs have been categorized based on the age of onset for symptoms (i.e. infantile, late infantile, juvenile, and adult) and the identity of the predisposing gene (Chabrol et al., 2013). ‘Batten’s disease’ selectively refers to juvenile NCL (JNCL) form; however, some have used the term to refer to all NCL forms collectively. The neurodegenerative disorder, which is progressive and can be fatal, typically occurs during childhood and there is no cure presently. Approximately 2-4 per 100,000 children are affected in the U. S. while the frequency may be higher (~1/12,500 people) in some populations.
To date, 14 genetically distinct NCL forms have been identified (Chabrol et al., 2013; Anu et al., 2009). ‘Infantile neuronal ceroid’ (INCL) is associated with CLN gene encoding PPT1 (palmitoyl-protein thioesterase 1) that functions as a lysosomal enzyme. Late infantile NCL (LINCL) with the life expectancy of 8-12 yrs is linked to CLN2 gene, which encodes the lysosomal enzyme TPP1 (tripeptidyl-peptidase 1). Variant of the late infantile NCL is caused by the disruption of CLN6 gene, which encodes the transmembrane protein of endoplasmic reticulum CLN6 (linclin). Another variant of late infantile NCL is associated with the CLN7 gene encoding MFSD8 (‘major facilitator superfamily domain containing eight’), a lysosomal transmembrane protein. ‘Juvenile NCL’ (JNCL) occurs due to the inactivation of CLN3 gene, which encodes a lysosomal transmembrane protein similar to SLC (solute carrier) transporters. Accurate diagnosis of NCL requires the determination of the mutated gene and the resultant dysfunctional protein (for genetic counseling).
Most NCLs exhibit autosomal recessive mode of inheritance. The disease is manifested if both alleles of the predisposing gene inherited from parents are defective. Though a carrier may not develop symptoms, the individual is capable of passing the defective allele to a descendent. An exception may be found in Kufs disease, a distinct form of NCL, whose symptoms appear around age 30 with a limited survival of less than 15 years after the onset. Its type A is linked to PPT1 and CLN6 genes, while type B (autosomal dominant) is associated with CTSF (cathepsin F) and DNAJC5 (‘DnaJ homolog subfamily C member 5’) genes (Benitez et al., 2011). Cathepsin is involved in proteolytic degradation in lysosomes.
Batten’s disease belongs to a group of disorders known as ‘lysosomal storage disease’, which is comprised of ca. 50 distinct autosomal recessively inherited genetic disorders. The disease occurs due to the dysfunctional lysosomes, resulting in the intracellular accumulation of excess amount of undegraded substrates. The dysfunction is caused by a defect or insufficient level of the lysosomal enzymes (ex. cathepsin, alpha-glucosidase, acid phosphatase) for the degradation of carbohydrates, lipids, proteins, nucleic acids, etc. The failure to breakdown into smaller components could lead to the death of the affected cells (ex. neurons) eventually. Lysosomes are also involved in autophagy (wherein autophagnosomes fuse with lysosomes to generate autolysosomes to degrade defective intracellular proteins, damaged organelles, pathogenic microbes, etc.), which has been associated with aging-associated disorders, heart disease, cancer, etc.
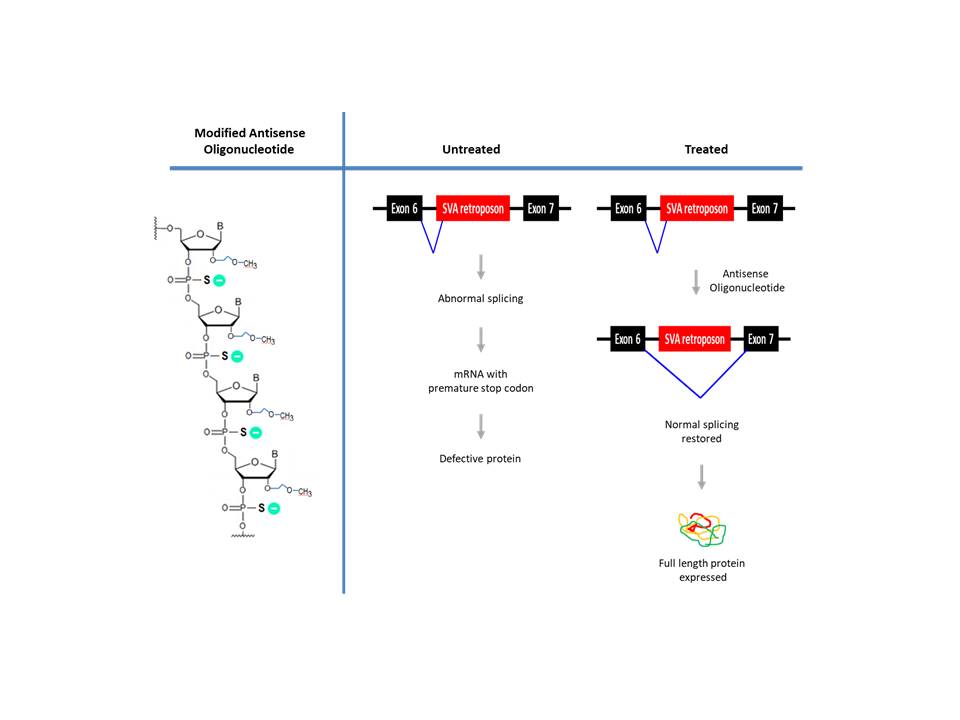
Batten’s disease remains a terminal disease with limited treatment options. In 2017, FDA approved the enzyme replacement therapy, Brineura, which involves intraventricularly administering recombinant human TPP1, the missing enzyme for NCL patients with defective CLN2 gene (Johnson et al., 2019). More recently, an mRNA-modulating treatment utilizing antisense oligonucleotides was reported though applicable for only a single patient (Kim et al., 2019). A 6-year-old girl with worsening symptoms, which included seizures, ataxia, developmental regression, deteriorating vision with mild cerebral/cerebellar atrophy, was diagnosed with Batten’s disease. Genetic analysis revealed that she was heterozygous for a pathogenic mutation in the MFSD8 (CLN7) gene. The whole genome sequencing showed that the other MFSD8 allele (both the patient and mother) contains the SVA (a composite of SINE-VNTR-Alu) retroposon inserted in intron 6. The retroposon caused exon 6 to mis-splice into the cryptic splice-acceptor site, ‘i6.SA’, located upstream of the retroposon, resulting in a faulty transcript with premature translational termination.
To correct the altered splicing, investigators at the Harvard University-affiliated Boston Children’s Hospital designed Milasen, a 22 bp antisense oligonucleotide, to block the aberrant ‘i6.SA’ splice acceptor site or exonic splicing enhancer (ESE) elements nearby. To increase stability, the oligonucleotide consisted of 2′-O-methoxyethyl (MOE) ribonucleotides with phosphorothioate internucleotide linkages. How the modified oligonucleotides were uptaken by neural cells is not clear though phsophorothioate-modified oligonucleotide is known to interact with several receptors (ex. stabilin receptor in the liver) (Miller et al., 2017). Milasen increased the ratio of normal-to-mutant splicing 2.5 to 3-fold in patient derived fibroblasts. The investigational drug Milasen was administered intrathecally to distribute to the brain via cerebrospinal fluid. During the treatment (~1 year), no serious adverse events occurred according to the authors. Though it did not cure, a notable reduction in the intensity and frequency of seizures was reported (Kim et al., 2019).
Critical to the above undertaking was the ability to synthesize oligonucleotides expeditiously. Bio-Synthesis, Inc. specializes in oligonucleotide modification and provides an extensive array of chemically modified nucleoside analogues (over ~200) including bridged nucleic acid (BNA). It recently acquired a license from BNA Inc. of Osaka, Japan, for the manufacturing and distribution of BNANC, a third generation of BNA oligonucleotides. To meet the demands of therapeutic application, its oligonucleotide products are approaching GMP grade. Bio-Synthesis, Inc. has recently entered into collaborative agreement with Bind Therapeutics, Inc. to synthesize miR-21 blocker using BNA for triple negative breast cancer. The BNA technology that we offer provides superior, unequalled advantages in base stacking, binding affinity, aqueous solubility and nuclease resistance. It also improves the formation of duplexes and triplexes by reducing the repulsion between the negatively charged phosphates of the oligonucleotide backbone. Its single-mismatch discriminating power was especially useful for diagnosis (ex. FISH using DNA probe). More importantly, BNA oligonucleotide exhibits lesser toxicity than other modified nucleotides for clinical application.
https://www.biosyn.com/oligonucleotide-modification-services.aspx
References
Anu J,Thomas B. "Neuronal ceroid lipofuscinoses". (2009). Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1793: 697–709. PMID 19084560 doi:10.1016/j.bbamcr.2008.11.004.
Benitez BA, Alvarado D, Cai Y, Mayo K, Chakraverty S, Norton J, Morris JC, Sands MS, Goate A, Cruchaga C. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. (2011) PLoS One. 6(11):e26741. PMID: 22073189 doi: 10.1371/journal.pone.0026741
Chabrol B, Caillaud C, Minassian B. Neuronal ceroid lipofuscinoses. (2013) Handbk Clin Neurol. 113:1701-6. PMID: 23622391 doi: 10.1016/B978-0-444-59565-2.00038-1.
Johnson TB, Cain JT, White KA, Ramirez-Montealegre D, Pearce DA, Weimer JM. Therapeutic landscape for Batten disease: current treatments and future prospects. (2019) Nat Rev Neurol. 15:161-178. doi: 10.1038/s41582-019-0138-8.
Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. (2019) N Engl J Med. 381:1644-1652. PMID:31597037 doi: 10.1056/NEJMoa1813279. Epub 2019 Oct 9.
Miller CM, Tanowitz M, Donner AJ, Prakash TP, Swayze EE, Harris EN, Seth PP. Receptor-mediated uptake of phosphorothioate antisense oligonucleotides in different cell types of the liver. (2018) Nucleic Acid Ther. 28:119-127. PMID: 29425080 doi: 10.1089/nat.2017.0709